

TWEAK and RIPK1 mediate a second wave of cell death during AKI
- PMID: 29588419
- PMCID: PMC5910825
- DOI: 10.1073/pnas.1716578115
TWEAK and RIPK1 mediate a second wave of cell death during AKI
Erratum in
-
Correction for Martin-Sanchez et al., TWEAK and RIPK1 mediate a second wave of cell death during AKI.Proc Natl Acad Sci U S A. 2018 May 15;115(20):E4731. doi: 10.1073/pnas.1806694115. Epub 2018 May 7. Proc Natl Acad Sci U S A. 2018. PMID: 29735703 Free PMC article. No abstract available.
Abstract
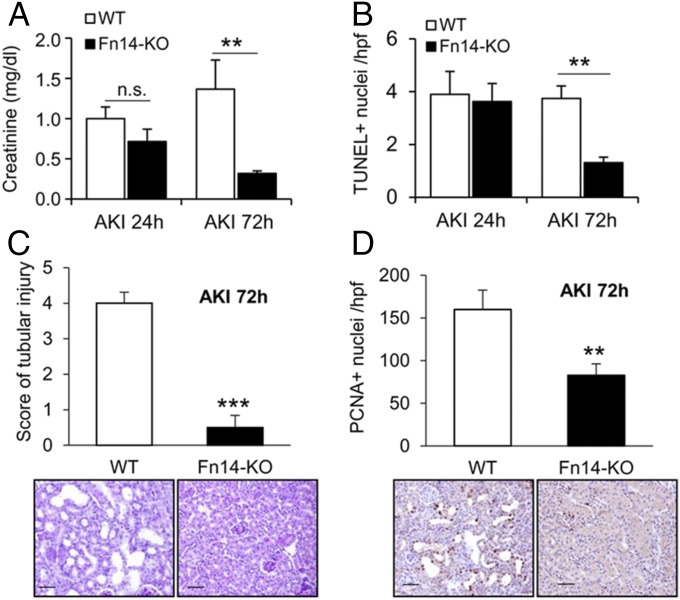
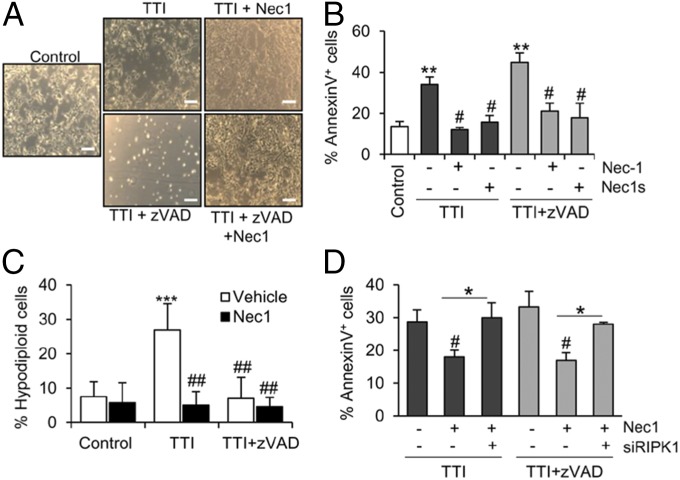
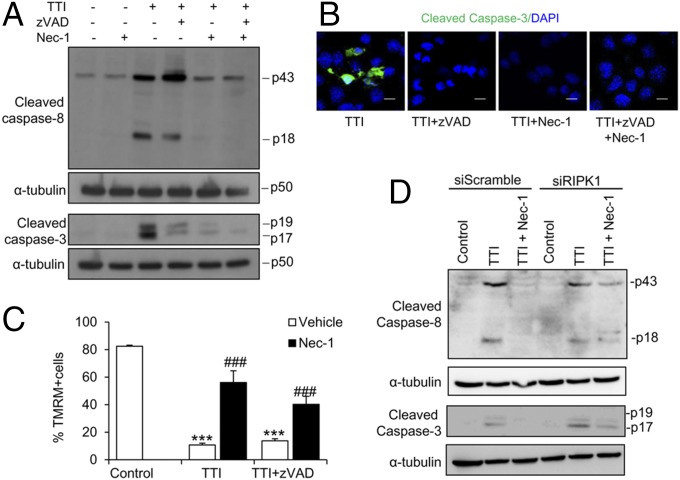
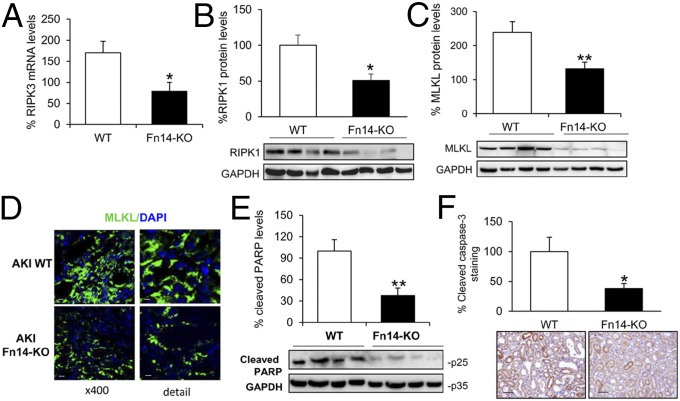
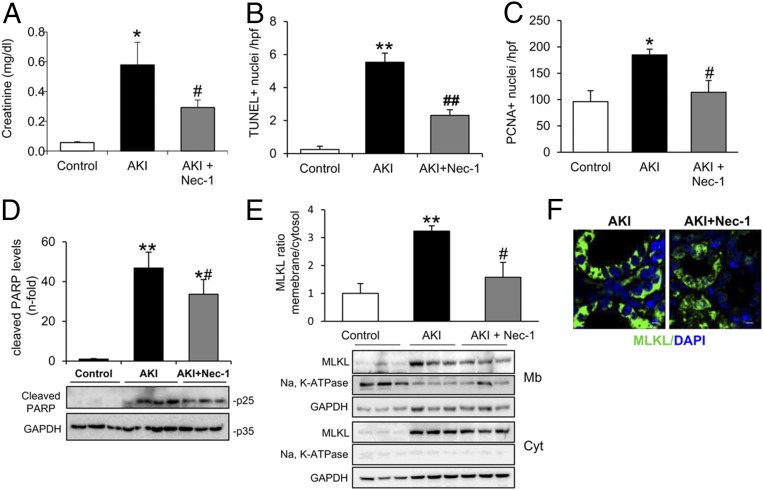
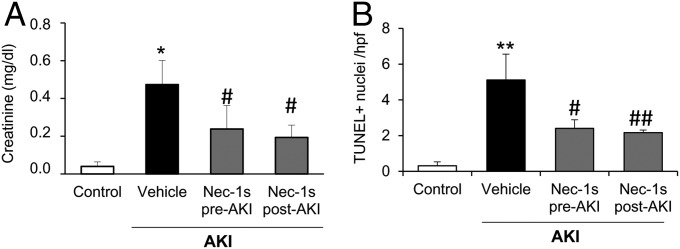
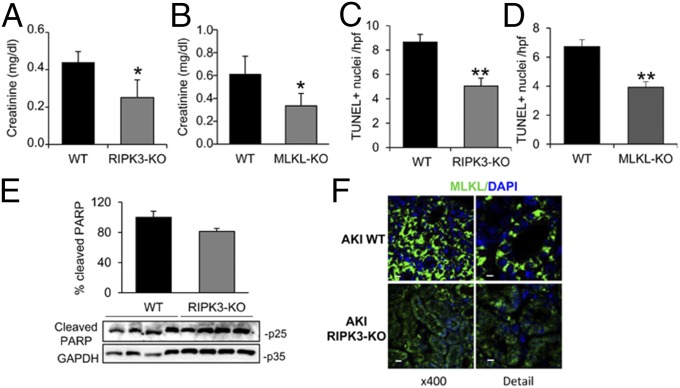
Acute kidney injury (AKI) is characterized by necrotic tubular cell death and inflammation. The TWEAK/Fn14 axis is a mediator of renal injury. Diverse pathways of regulated necrosis have recently been reported to contribute to AKI, but there are ongoing discussions on the timing or molecular regulators involved. We have now explored the cell death pathways induced by TWEAK/Fn14 activation and their relevance during AKI. In cultured tubular cells, the inflammatory cytokine TWEAK induces apoptosis in a proinflammatory environment. The default inhibitor of necroptosis [necrostatin-1 (Nec-1)] was protective, while caspase inhibition switched cell death to necroptosis. Additionally, folic acid-induced AKI in mice resulted in increased expression of Fn14 and necroptosis mediators, such as receptor-interacting protein kinase 1 (RIPK1), RIPK3, and mixed lineage domain-like protein (MLKL). Targeting necroptosis with Nec-1 or by genetic RIPK3 deficiency and genetic Fn14 ablation failed to be protective at early time points (48 h). However, a persistently high cell death rate and kidney dysfunction (72-96 h) were dependent on an intact TWEAK/Fn14 axis driving necroptosis. This was prevented by Nec-1, or MLKL, or RIPK3 deficiency and by Nec-1 stable (Nec-1s) administered before or after induction of AKI. These data suggest that initial kidney damage and cell death are amplified through recruitment of inflammation-dependent necroptosis, opening a therapeutic window to treat AKI once it is established. This may be relevant for clinical AKI, since using current diagnostic criteria, severe injury had already led to loss of renal function at diagnosis.
Keywords: AKI; Fn14; RIPK1; TWEAK; cell death.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
