

GENEASE: real time bioinformatics tool for multi-omics and disease ontology exploration, analysis and visualization
- PMID: 29590301
- PMCID: PMC6137982
- DOI: 10.1093/bioinformatics/bty182
GENEASE: real time bioinformatics tool for multi-omics and disease ontology exploration, analysis and visualization
Abstract
Motivation: Advances in high-throughput sequencing technologies have made it possible to generate multiple omics data at an unprecedented rate and scale. The accumulation of these omics data far outpaces the rate at which biologists can mine and generate new hypothesis to test experimentally. There is an urgent need to develop a myriad of powerful tools to efficiently and effectively search and filter these resources to address specific post-GWAS functional genomics questions. However, to date, these resources are scattered across several databases and often lack a unified portal for data annotation and analytics. In addition, existing tools to analyze and visualize these databases are highly fragmented, resulting researchers to access multiple applications and manual interventions for each gene or variant in an ad hoc fashion until all the questions are answered.
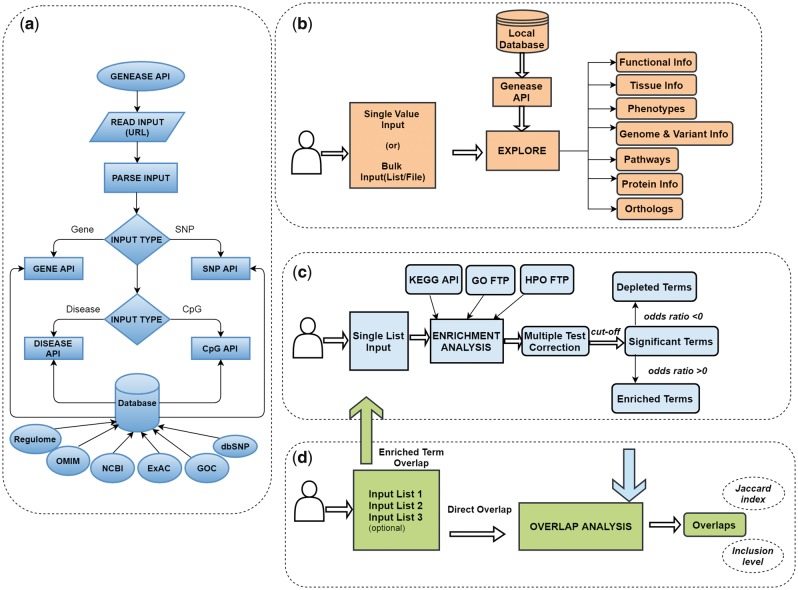
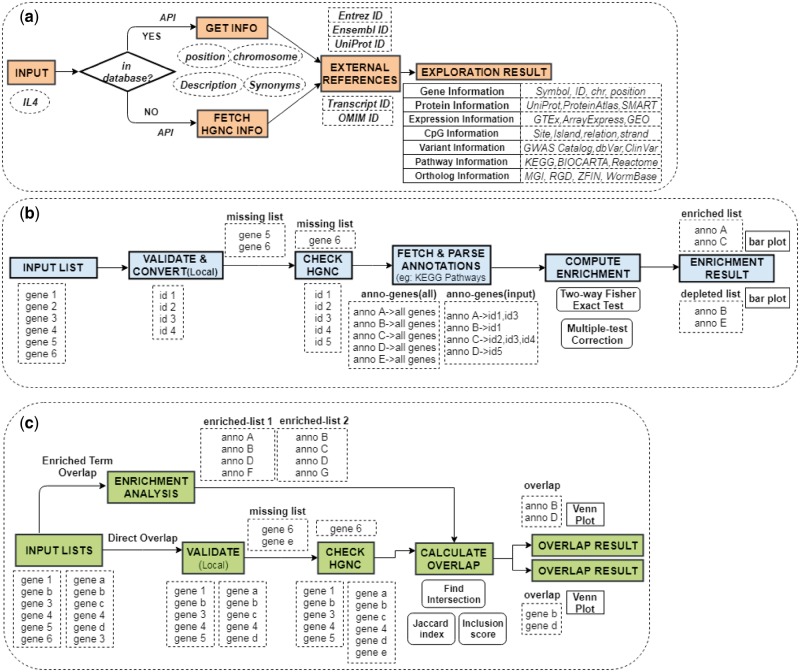
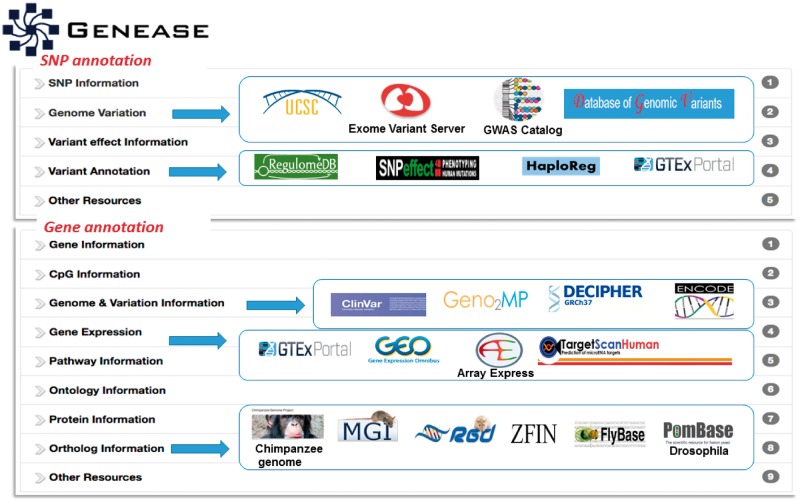
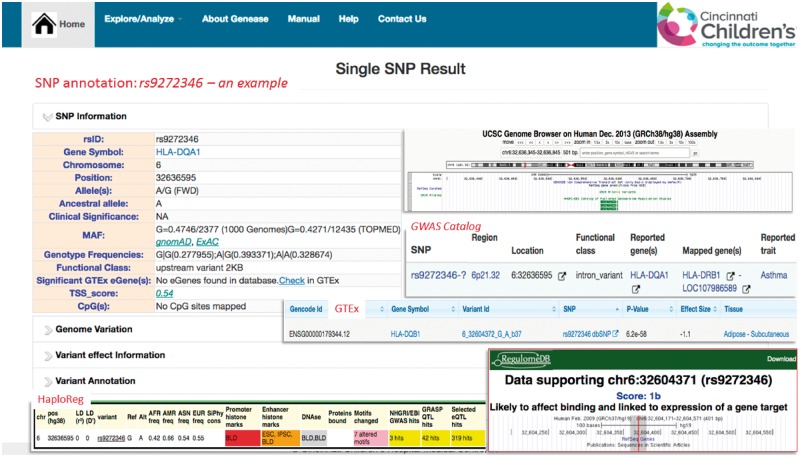
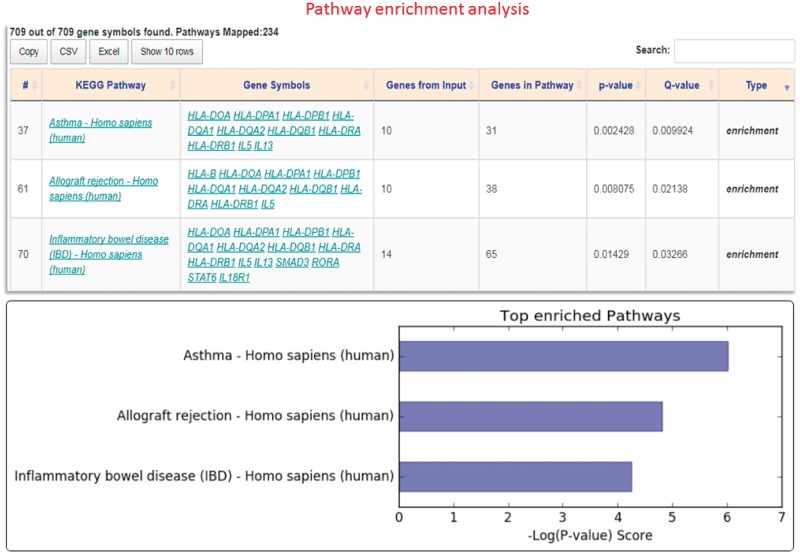
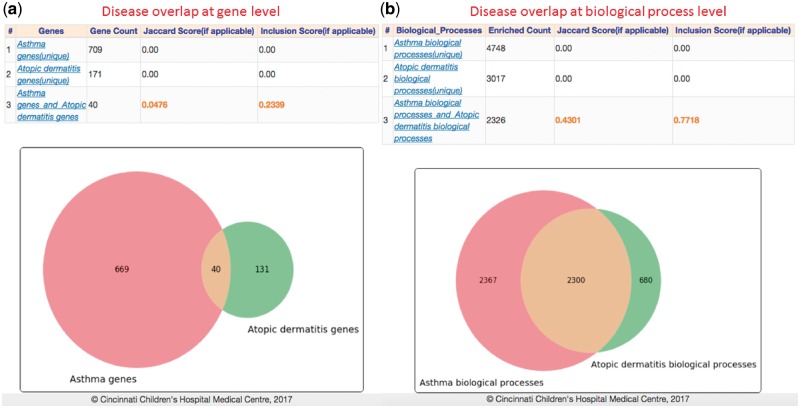
Results: In this study, we present GENEASE, a web-based one-stop bioinformatics tool designed to not only query and explore multi-omics and phenotype databases (e.g. GTEx, ClinVar, dbGaP, GWAS Catalog, ENCODE, Roadmap Epigenomics, KEGG, Reactome, Gene and Phenotype Ontology) in a single web interface but also to perform seamless post genome-wide association downstream functional and overlap analysis for non-coding regulatory variants. GENEASE accesses over 50 different databases in public domain including model organism-specific databases to facilitate gene/variant and disease exploration, enrichment and overlap analysis in real time. It is a user-friendly tool with point-and-click interface containing links for support information including user manual and examples.
Availability and implementation: GENEASE can be accessed freely at http://research.cchmc.org/mershalab/GENEASE/login.html.
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures
References
-
- Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc., 57, 12.
-
- Eck N.J.v., Waltman L. (2009), How to normalize co-occurrence data? An analysis of some well-known similarity measures, In: ERIM report series research in management, Erasmus Research Institute of Management, (January 2009), p. 42.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
