

Mek1Y130C mice recapitulate aspects of human cardio-facio-cutaneous syndrome
- PMID: 29590634
- PMCID: PMC5897723
- DOI: 10.1242/dmm.031278
Mek1Y130C mice recapitulate aspects of human cardio-facio-cutaneous syndrome
Abstract
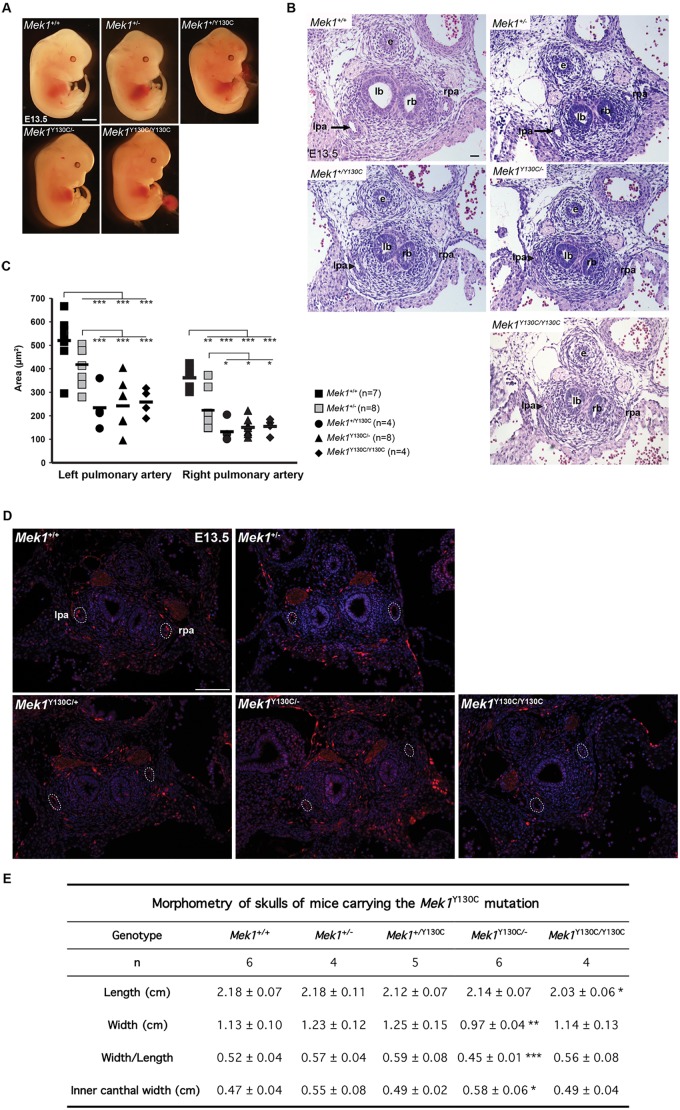
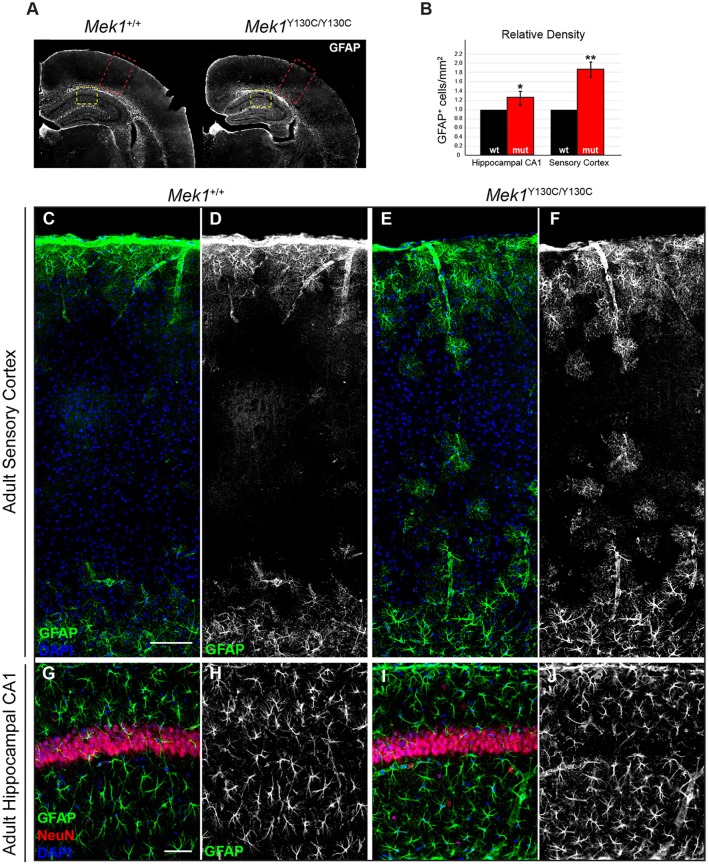
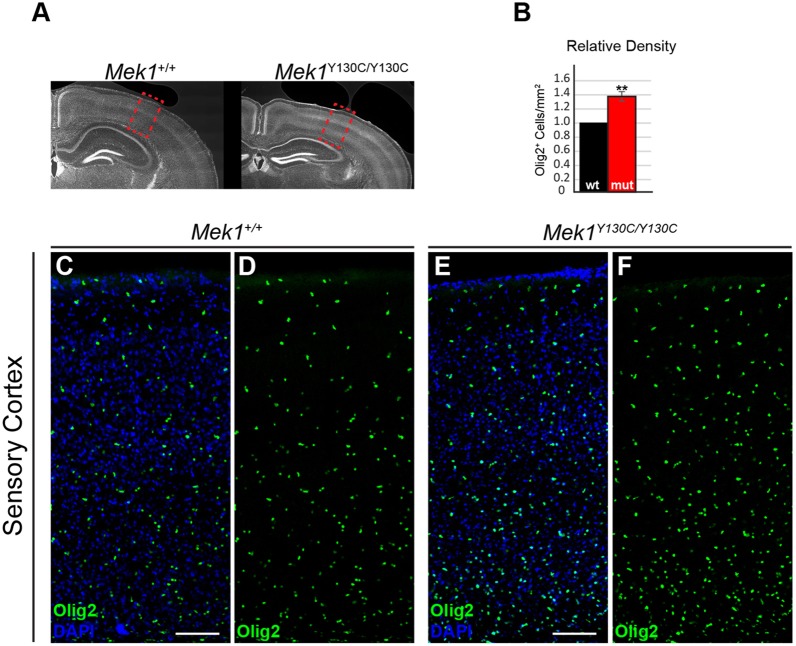
The RAS/MAPK signaling pathway is one of the most investigated pathways, owing to its established role in numerous cellular processes and implication in cancer. Germline mutations in genes encoding members of the RAS/MAPK pathway also cause severe developmental syndromes collectively known as RASopathies. These syndromes share overlapping characteristics, including craniofacial dysmorphology, cardiac malformations, cutaneous abnormalities and developmental delay. Cardio-facio-cutaneous syndrome (CFC) is a rare RASopathy associated with mutations in BRAF, KRAS, MEK1 (MAP2K1) and MEK2 (MAP2K2). MEK1 and MEK2 mutations are found in ∼25% of the CFC patients and the MEK1Y130C substitution is the most common one. However, little is known about the origins and mechanisms responsible for the development of CFC. To our knowledge, no mouse model carrying RASopathy-linked Mek1 or Mek2 gene mutations has been reported. To investigate the molecular and developmental consequences of the Mek1Y130C mutation, we generated a mouse line carrying this mutation. Analysis of mice from a Mek1 allelic series revealed that the Mek1Y130C allele expresses both wild-type and Y130C mutant forms of MEK1. However, despite reduced levels of MEK1 protein and the lower abundance of MEK1 Y130C protein than wild type, Mek1Y130C mutants showed increased ERK (MAPK) protein activation in response to growth factors, supporting a role for MEK1 Y130C in hyperactivation of the RAS/MAPK pathway, leading to CFC. Mek1Y130C mutant mice exhibited pulmonary artery stenosis, cranial dysmorphia and neurological anomalies, including increased numbers of GFAP+ astrocytes and Olig2+ oligodendrocytes in regions of the cerebral cortex. These data indicate that the Mek1Y130C mutation recapitulates major aspects of CFC, providing a new animal model to investigate the physiopathology of this RASopathy. This article has an associated First Person interview with the first author of the paper.
Keywords: Cardio-facio-cutaneous syndrome; MEK1 Y130C mutation; Mouse model; Neurological defects; Pulmonary artery stenosis; RAS/MAPK pathway.
© 2018. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interestsThe authors declare no competing or financial interests.
Figures
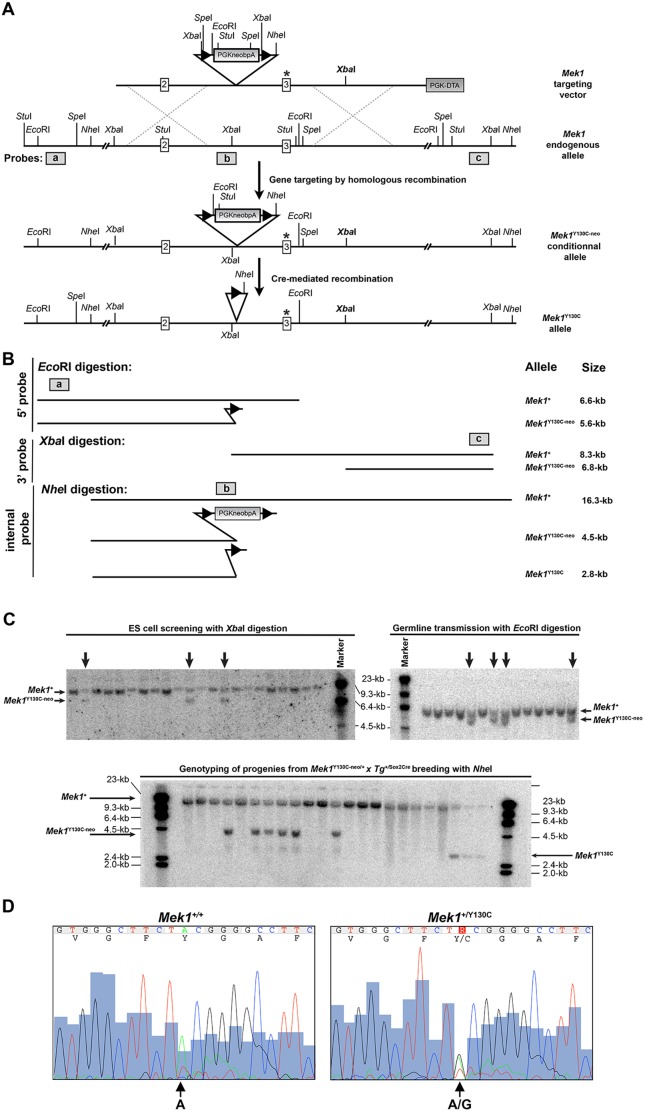
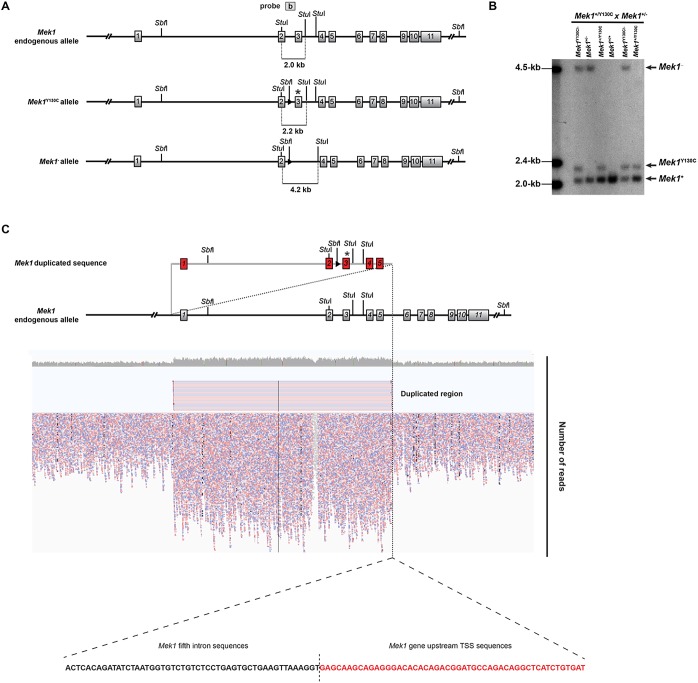
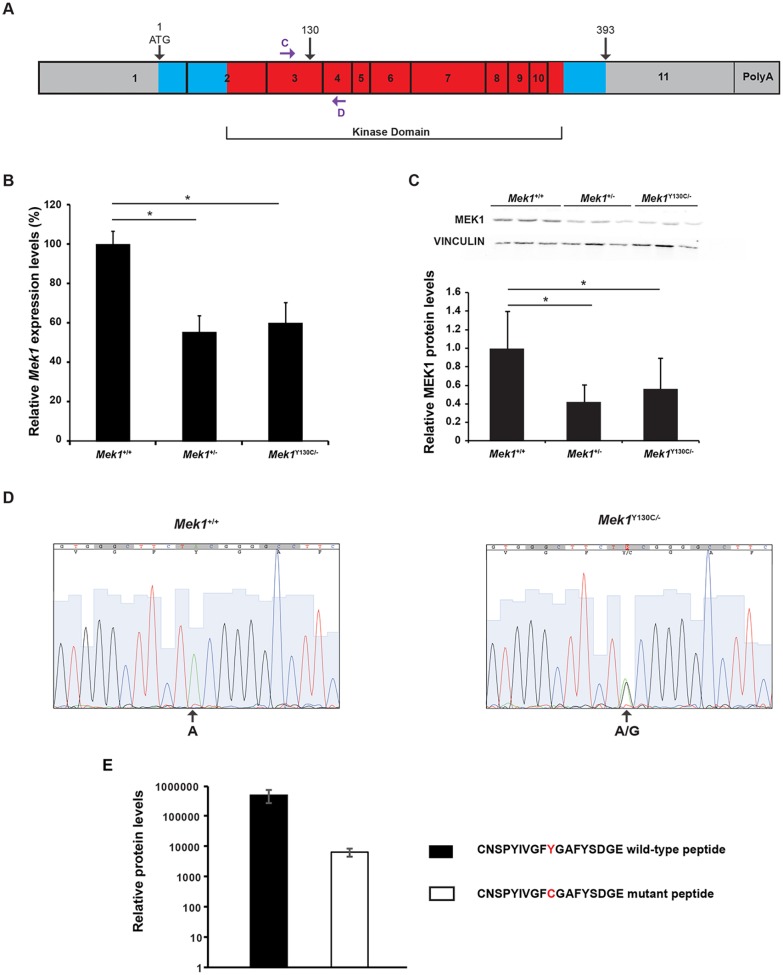
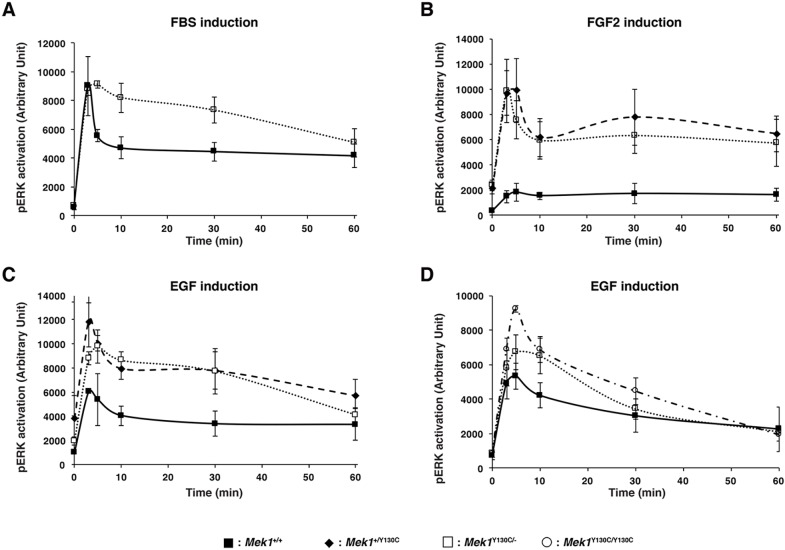
References
-
- Andreadi C., Cheung L.-K., Giblett S., Patel B., Jin H., Mercer K., Kamata T., Lee P., Williams A., McMahon M. et al. (2012). The intermediate-activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev. 26, 1945-1958. 10.1101/gad.193458.112 - DOI - PMC - PubMed
-
- Araki T., Mohi M. G., Ismat F. A., Bronson R. T., Williams I. R., Kutok J. L., Yang W., Pao L. I., Gilliland D. G., Epstein J. A. et al. (2004). Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat. Med. 10, 849-857. 10.1038/nm1084 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
