

Emerging recombinant noroviruses identified by clinical and waste water screening
- PMID: 29593246
- PMCID: PMC5874246
- DOI: 10.1038/s41426-018-0047-8
Emerging recombinant noroviruses identified by clinical and waste water screening
Abstract
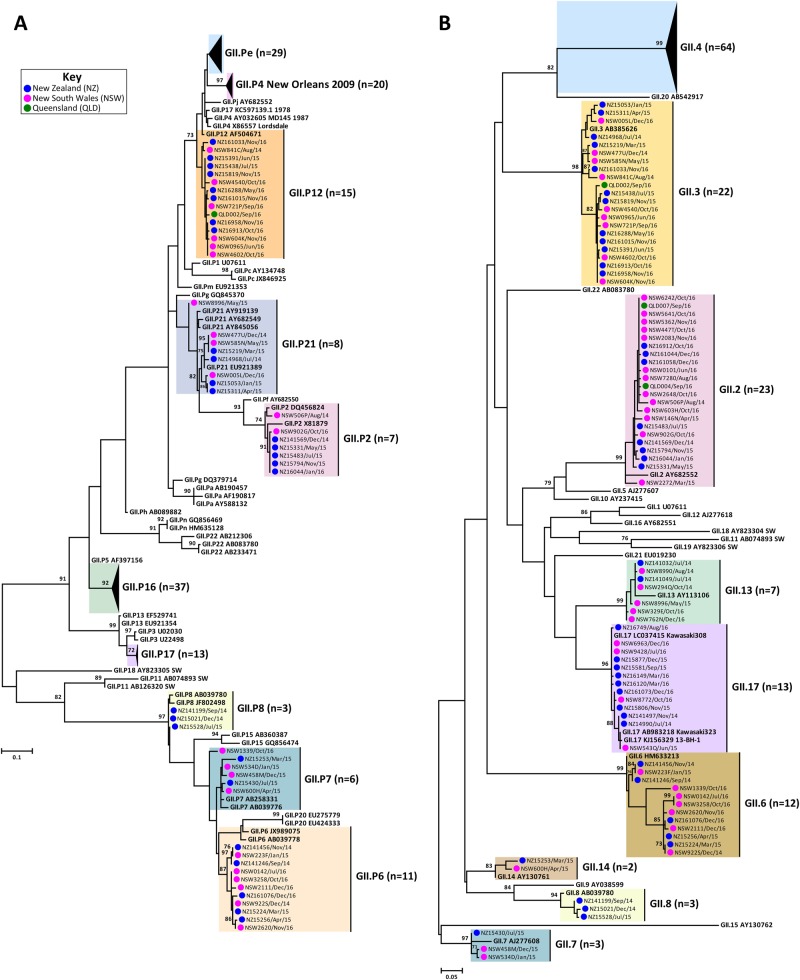
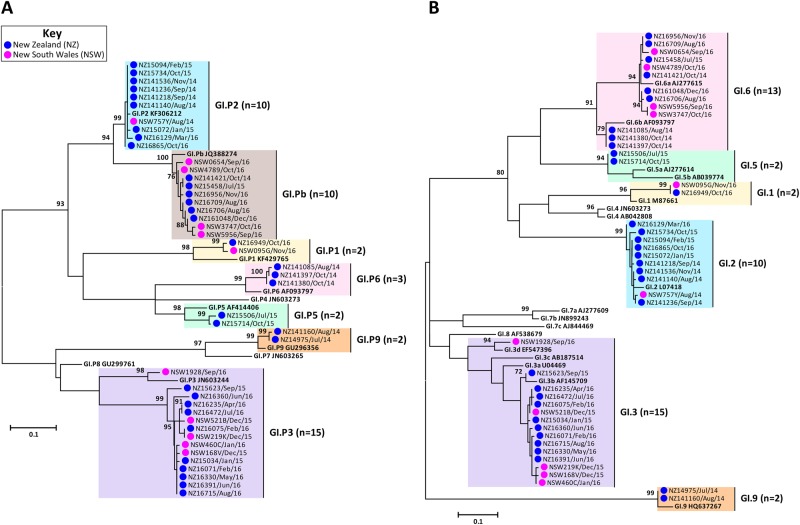
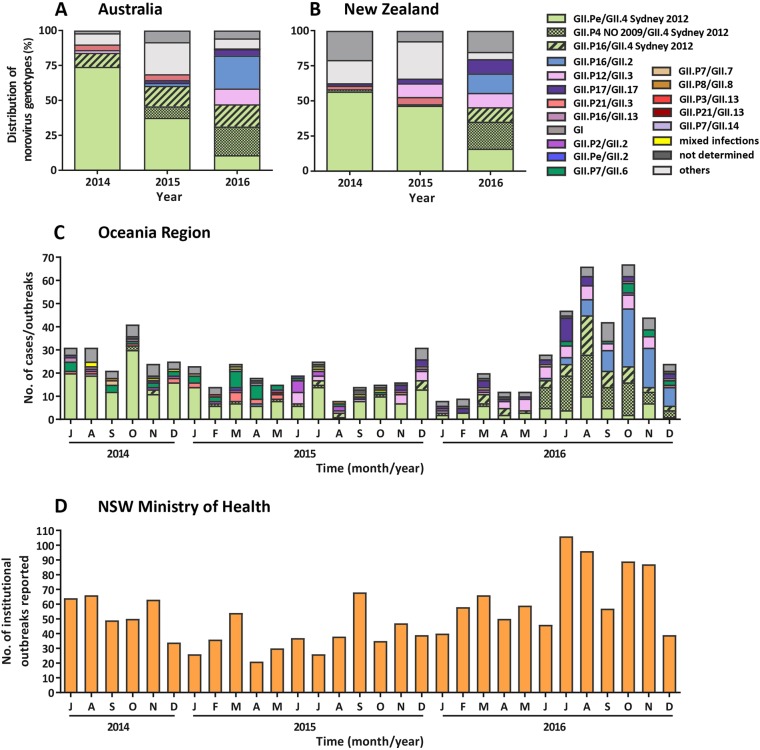
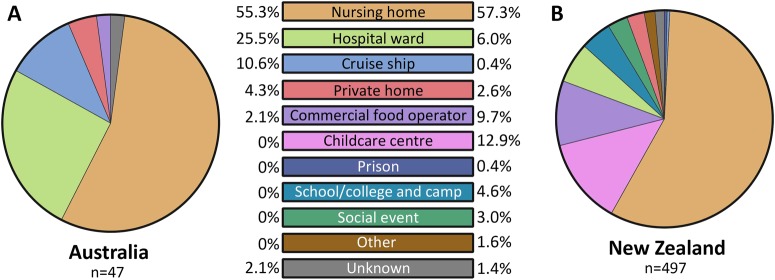
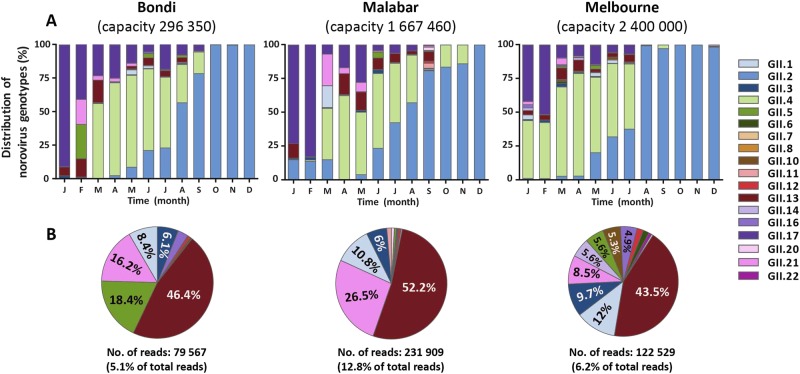
Norovirus is estimated to cause 677 million annual cases of gastroenteritis worldwide, resulting in 210,000 deaths. As viral gastroenteritis is generally self-limiting, clinical samples for epidemiological studies only partially represent circulating noroviruses in the population and is biased towards severe symptomatic cases. As infected individuals from both symptomatic and asymptomatic cases shed viruses into the sewerage system at a high concentration, waste water samples are useful for the molecular epidemiological analysis of norovirus genotypes at a population level. Using Illumina MiSeq and Sanger sequencing, we surveyed circulating norovirus within Australia and New Zealand, from July 2014 to December 2016. Importantly, norovirus genomic diversity during 2016 was compared between clinical and waste water samples to identify potential pandemic variants, novel recombinant viruses and the timing of their emergence. Although the GII.4 Sydney 2012 variant was prominent in 2014 and 2015, its prevalence significantly decreased in both clinical and waste water samples over 2016. This was concomitant with the emergence of multiple norovirus strains, including twoGII.4 Sydney 2012 recombinant viruses, GII.P4 New Orleans 2009/GII.4 Sydney 2012 and GII.P16/GII.4 Sydney 2012, along with three other emerging strains GII.17, GII.P12/GII.3 and GII.P16/GII.2. This is unusual, as a single GII.4 pandemic variant is generally responsible for 65-80% of all human norovirus infections at any one time and predominates until it is replaced by a new pandemic variant. In sumary, this study demonstrates the combined use of clinical and wastewater samples provides a more complete picture of norovirus circulating within the population.
Conflict of interest statement
JHL’s Water Research Australia scholarship was funded by Melbourne Water Corporation. In addition, this work was partly funded by Melbourne Water Corporation. All other authors declare that they have no conflict of interests.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials