

EP4 as a Therapeutic Target for Aggressive Human Breast Cancer
- PMID: 29596308
- PMCID: PMC5979567
- DOI: 10.3390/ijms19041019
EP4 as a Therapeutic Target for Aggressive Human Breast Cancer
Abstract
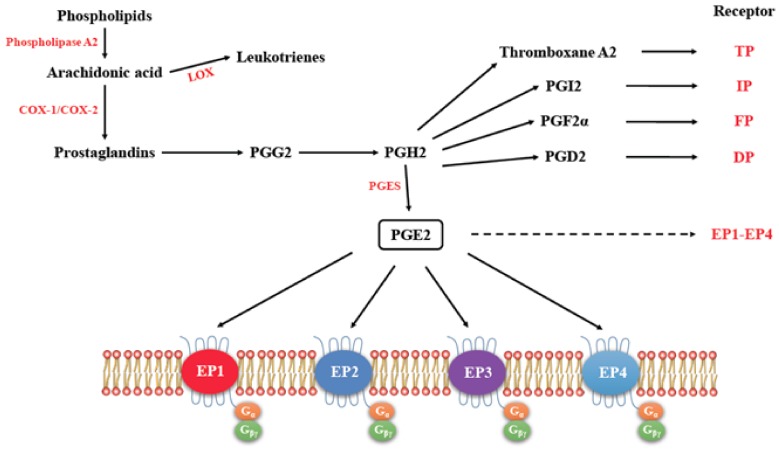
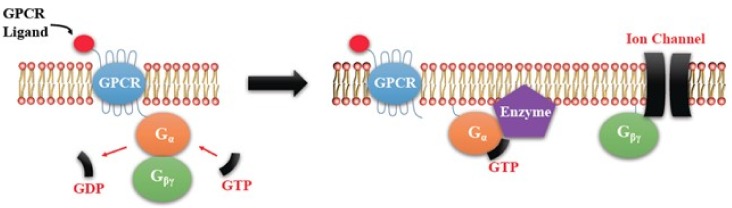
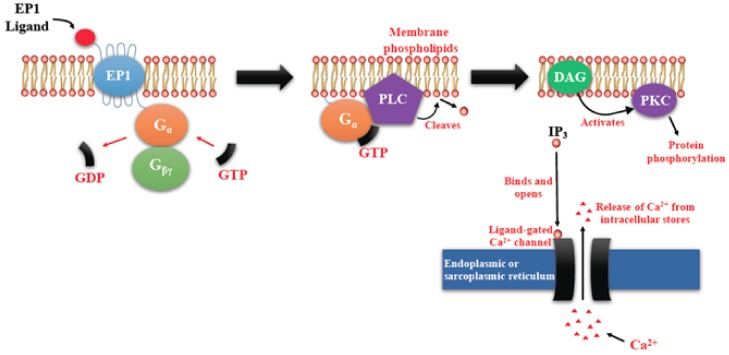
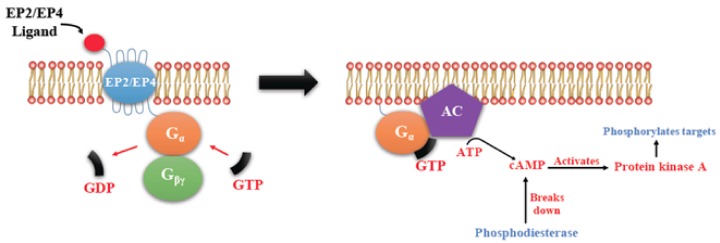
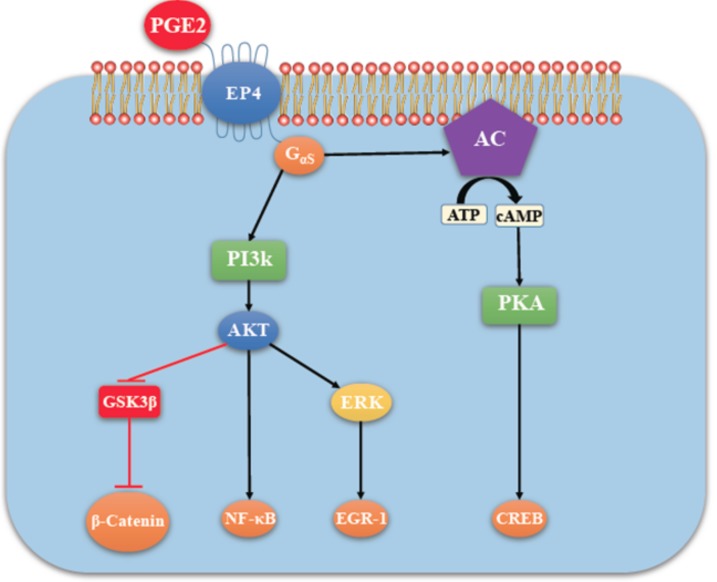
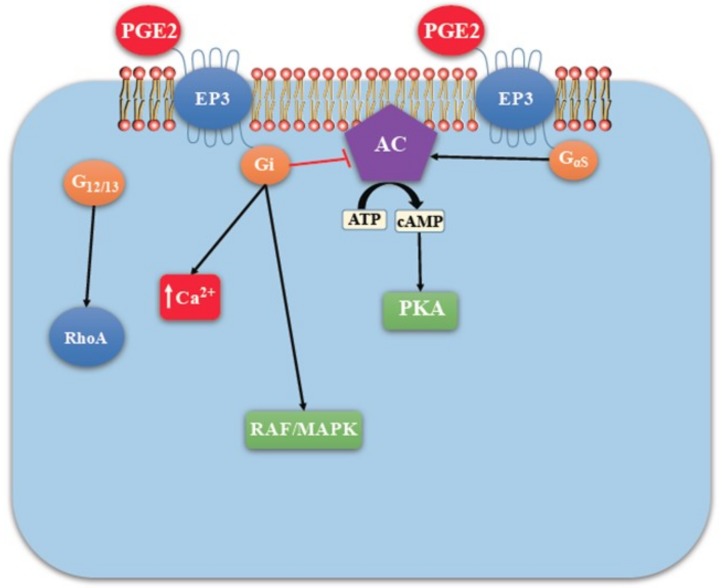
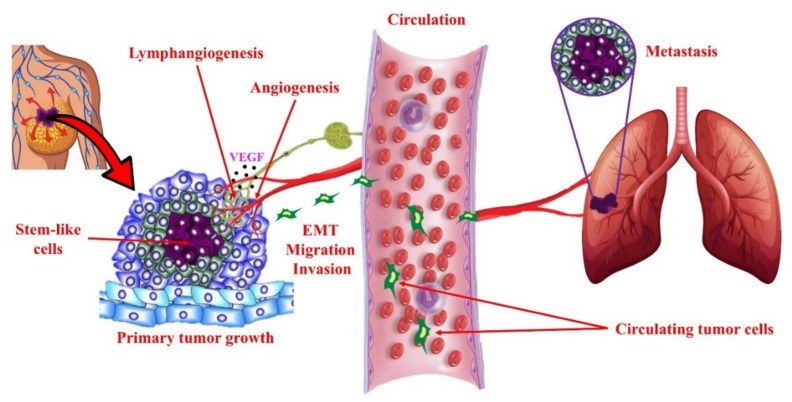
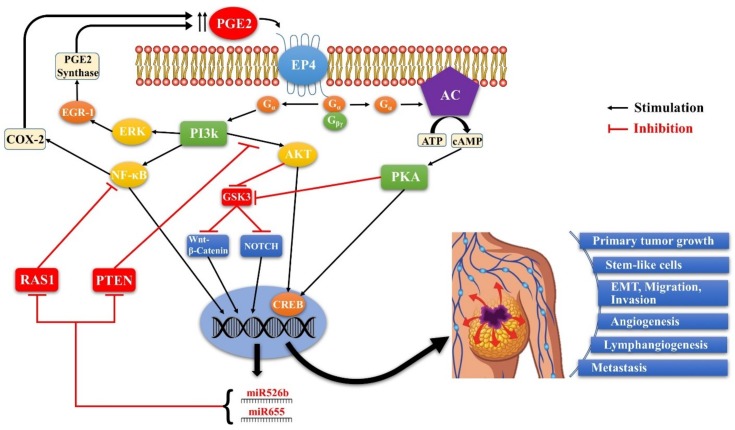
G-protein-coupled receptors (GPCRs, also called seven-transmembrane or heptahelical receptors) are a superfamily of cell surface receptor proteins that bind to many extracellular ligands and transmit signals to an intracellular guanine nucleotide-binding protein (G-protein). When a ligand binds, the receptor activates the attached G-protein by causing the exchange of Guanosine-5'-triphosphate (GTP) for guanosine diphosphate (GDP). They play a major role in many physiological functions, as well as in the pathology of many diseases, including cancer progression and metastasis. Only a few GPCR members have been exploited as targets for developing drugs with therapeutic benefit in cancer. Present review briefly summarizes the signaling pathways utilized by the EP (prostaglandin E receptor) family of GPCR, their physiological and pathological roles in carcinogenesis, with special emphasis on the roles of EP4 in breast cancer progression. We make a case for EP4 as a promising newer therapeutic target for treating breast cancer. We show that an aberrant over-expression of cyclooxygenase (COX)-2, which is an inflammation-associated enzyme, occurring in 40-50% of breast cancer patients leads to tumor progression and metastasis due to multiple cellular events resulting from an increased prostaglandin (PG) E2 production in the tumor milieu. They include inactivation of host anti-tumor immune cells, such as Natural Killer (NK) and T cells, increased immuno-suppressor function of tumor-associated macrophages, promotion of tumor cell migration, invasiveness and tumor-associated angiogenesis, due to upregulation of multiple angiogenic factors including Vascular Endothelial Growth Factor (VEGF)-A, increased lymphangiogenesis (due to upregulation of VEGF-C/D), and a stimulation of stem-like cell (SLC) phenotype in cancer cells. All of these events were primarily mediated by activation of the Prostaglandin (PG) E receptor EP4 on tumor or host cells. We show that selective EP4 antagonists (EP4A) could mitigate all of these events tested with cells in vitro as well as in vivo in syngeneic COX-2 expressing mammary cancer bearing mice or immune-deficient mice bearing COX-2 over-expressing human breast cancer xenografts. We suggest that EP4A can avoid thrombo-embolic side effects of long term use of COX-2 inhibitors by sparing cardio-protective roles of PGI2 via IP receptor activation or PGE2 via EP3 receptor activation. Furthermore, we identified two COX-2/EP4 induced oncogenic and SLC-stimulating microRNAs-miR526b and miR655, one of which (miR655) appears to be a potential blood biomarker in breast cancer patients for monitoring SLC-ablative therapies, such as with EP4A. We suggest that EP4A will likely produce the highest benefit in aggressive breast cancers, such as COX-2 expressing triple-negative breast cancers, when combined with other newer agents, such as inhibitors of programmed cell death (PD)-1 or PD-L1.
Keywords: COX-2; EP receptors; PGE2; angiogenesis; breast cancer; lymphangiogenesis; metastasis; microRNAs; stem-like cells; triple-negative breast cancer.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
