

Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines
- PMID: 29596782
- PMCID: PMC6075717
- DOI: 10.1016/j.cels.2018.03.002
Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines
Abstract
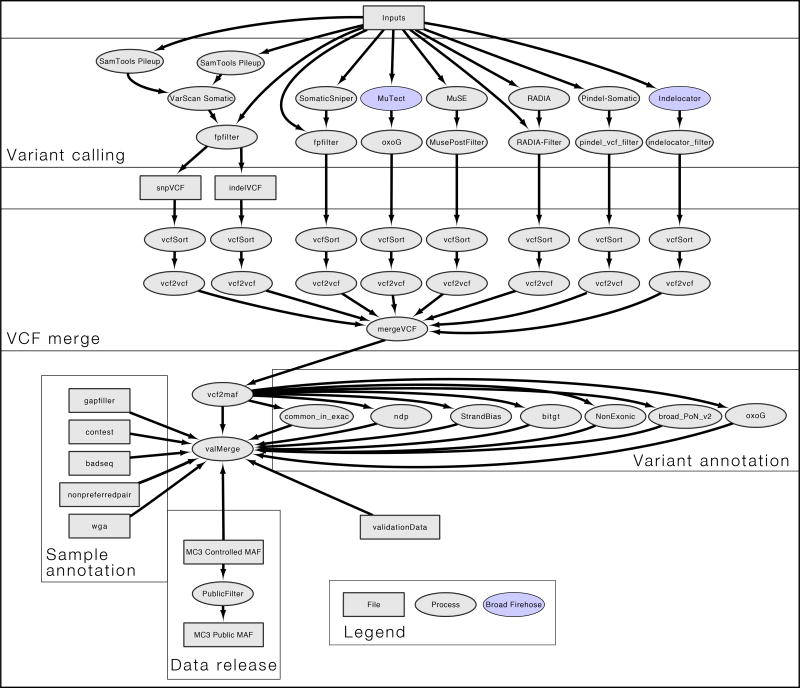
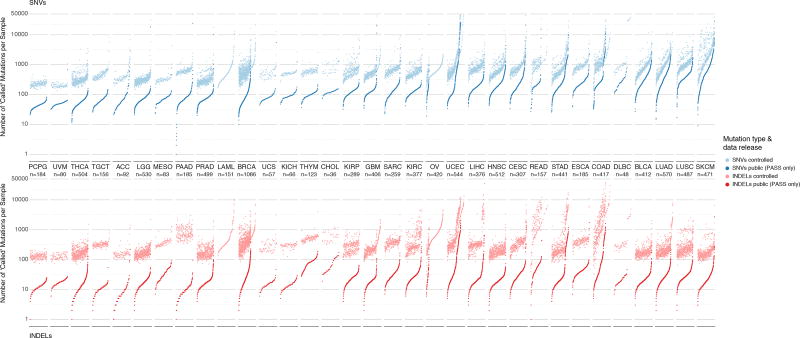
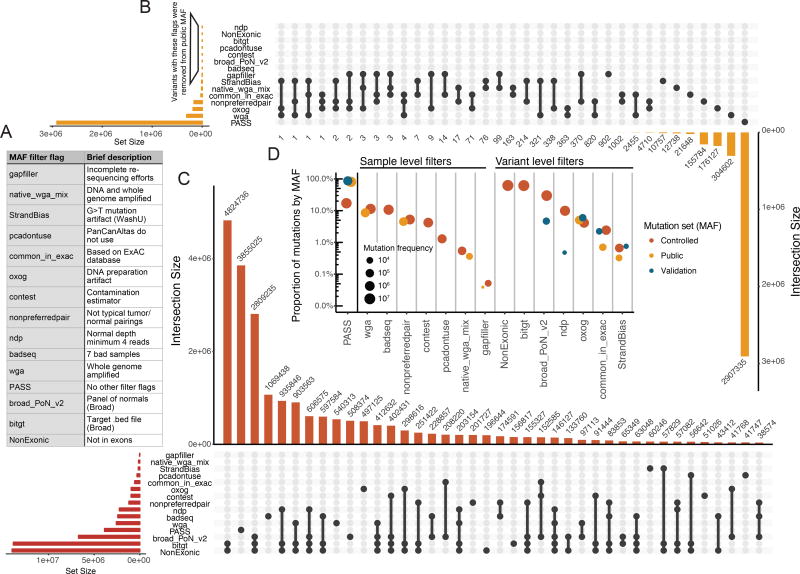
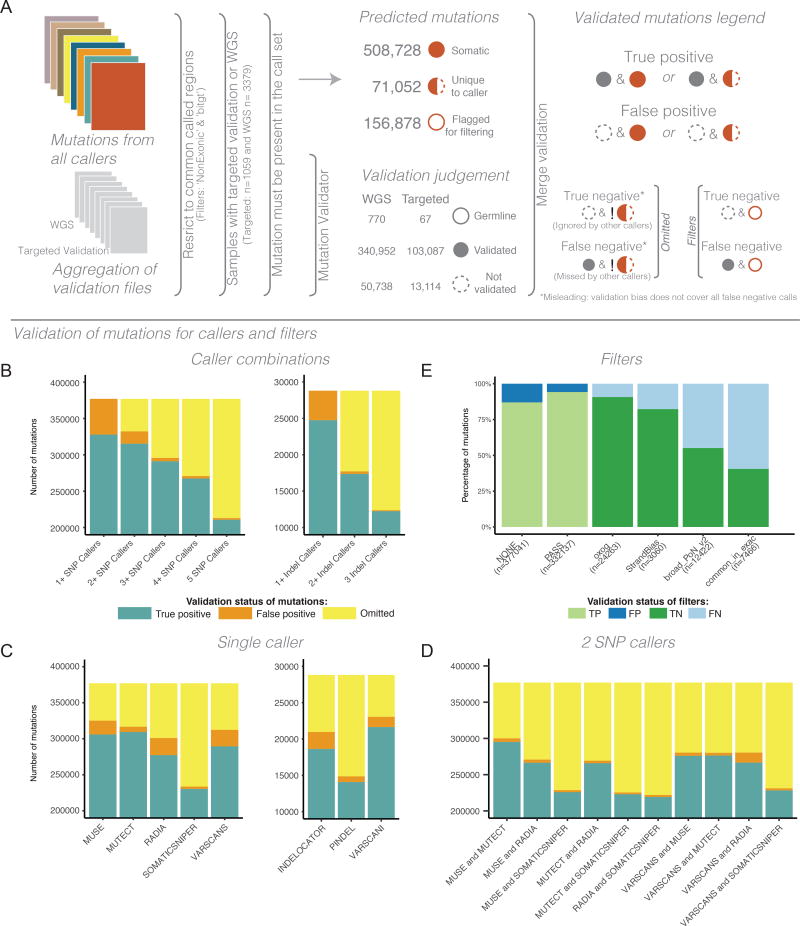
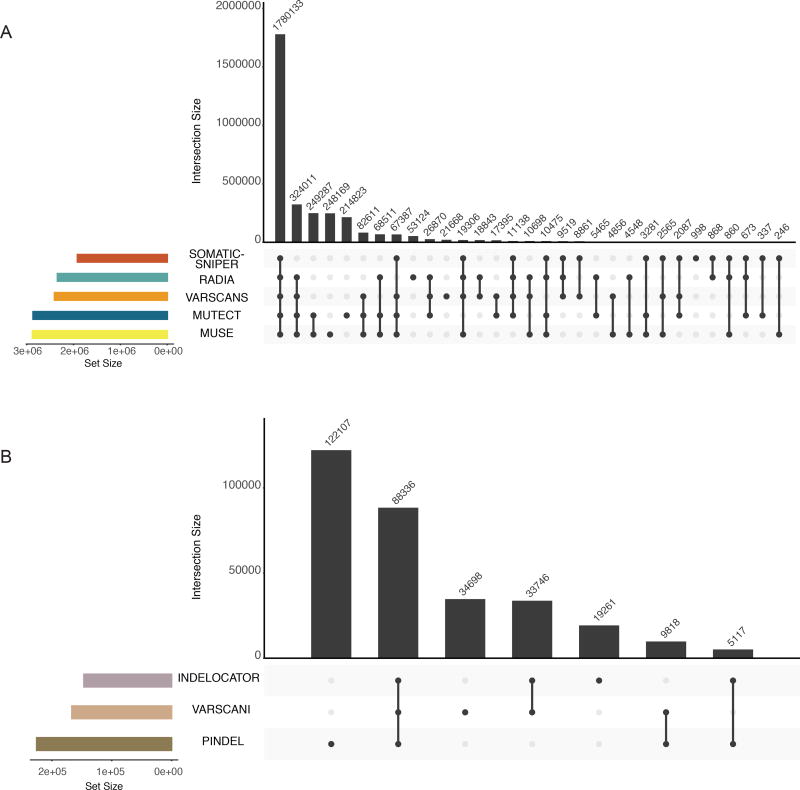
The Cancer Genome Atlas (TCGA) cancer genomics dataset includes over 10,000 tumor-normal exome pairs across 33 different cancer types, in total >400 TB of raw data files requiring analysis. Here we describe the Multi-Center Mutation Calling in Multiple Cancers project, our effort to generate a comprehensive encyclopedia of somatic mutation calls for the TCGA data to enable robust cross-tumor-type analyses. Our approach accounts for variance and batch effects introduced by the rapid advancement of DNA extraction, hybridization-capture, sequencing, and analysis methods over time. We present best practices for applying an ensemble of seven mutation-calling algorithms with scoring and artifact filtering. The dataset created by this analysis includes 3.5 million somatic variants and forms the basis for PanCan Atlas papers. The results have been made available to the research community along with the methods used to generate them. This project is the result of collaboration from a number of institutes and demonstrates how team science drives extremely large genomics projects.
Keywords: PanCanAtlas project; TCGA; large-scale; open science; pan-cancer; reproducible computing; somatic mutation calling.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Brunner AM, Graubert TA. Genomics in childhood acute myeloid leukemia comes of age. Nat. Med. 2018;24:7–9. - PubMed
-
- Campbell PJ, Getz G, Stuart JM, Korbel JO, Stein LD Net, - ICGC/TCGA Pan-Cancer Analysis of Whole Genomes. Pan-cancer analysis of whole genomes. bioRxiv 162784 2017
Publication types
MeSH terms
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- U24 CA143882/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- R01 CA183793/CA/NCI NIH HHS/United States
- R01 CA180778/CA/NCI NIH HHS/United States
- U24 CA210949/CA/NCI NIH HHS/United States
- R01 CA163722/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- U01 HG006517/HG/NHGRI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24 CA210969/CA/NCI NIH HHS/United States
- U54 HG007990/HG/NHGRI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA211006/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
