

Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing
- PMID: 29598823
- PMCID: PMC5877393
- DOI: 10.1186/s13059-018-1418-0
Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing
Abstract
Background: The multifaceted control of gene expression requires tight coordination of regulatory mechanisms at transcriptional and post-transcriptional level. Here, we studied the interdependence of transcription initiation, splicing and polyadenylation events on single mRNA molecules by full-length mRNA sequencing.
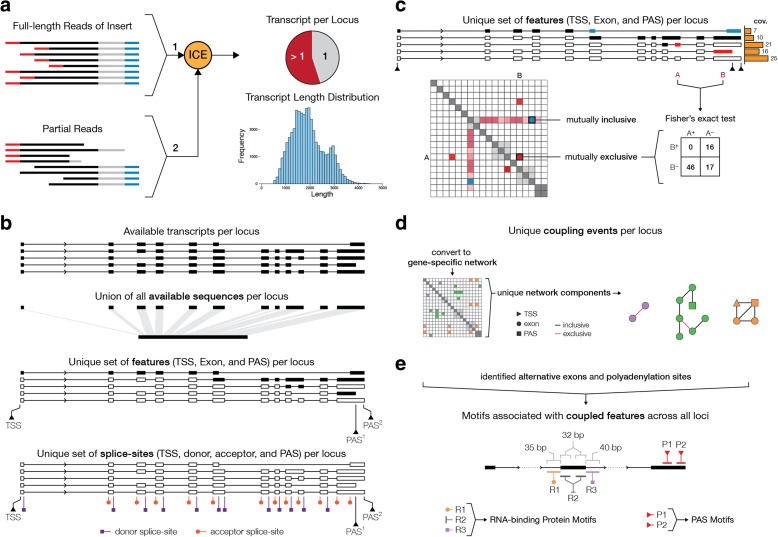
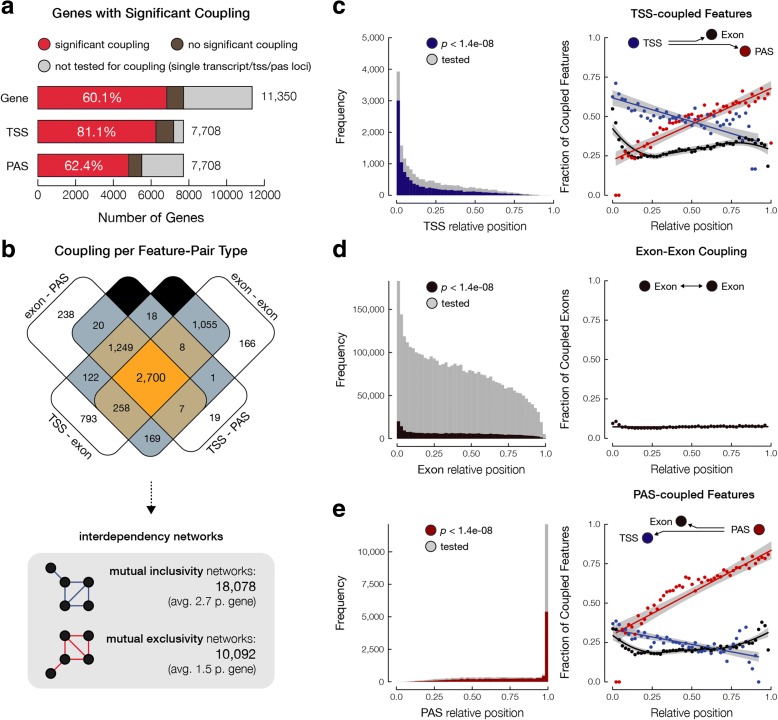
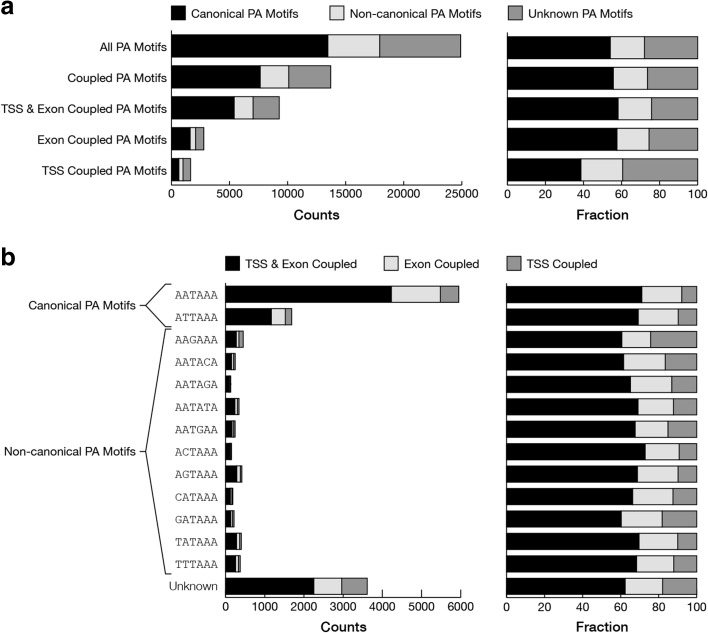
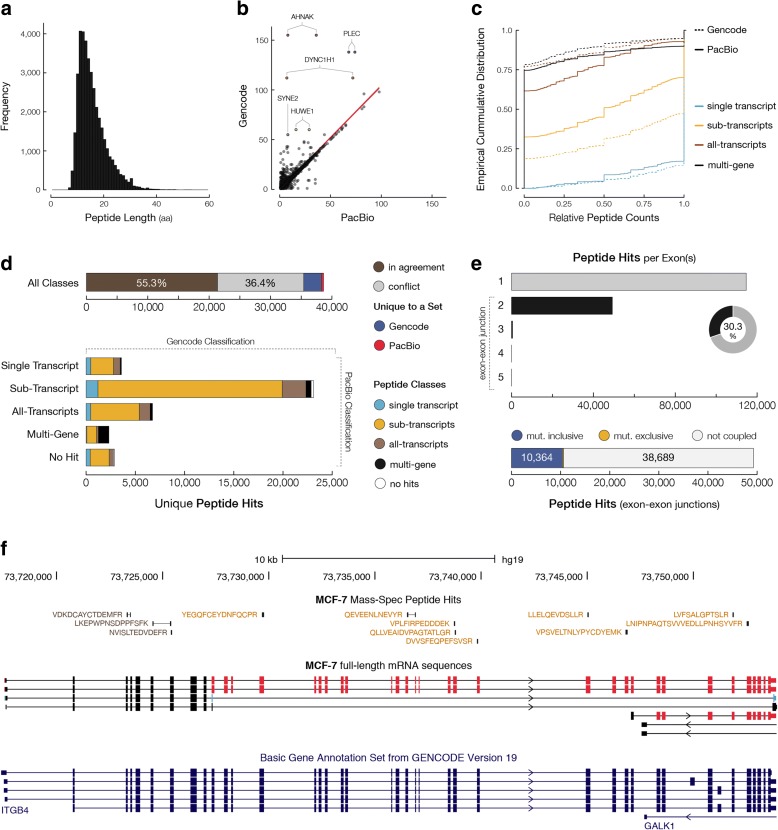
Results: In MCF-7 breast cancer cells, we find 2700 genes with interdependent alternative transcription initiation, splicing and polyadenylation events, both in proximal and distant parts of mRNA molecules, including examples of coupling between transcription start sites and polyadenylation sites. The analysis of three human primary tissues (brain, heart and liver) reveals similar patterns of interdependency between transcription initiation and mRNA processing events. We predict thousands of novel open reading frames from full-length mRNA sequences and obtained evidence for their translation by shotgun proteomics. The mapping database rescues 358 previously unassigned peptides and improves the assignment of others. By recognizing sample-specific amino-acid changes and novel splicing patterns, full-length mRNA sequencing improves proteogenomics analysis of MCF-7 cells.
Conclusions: Our findings demonstrate that our understanding of transcriptome complexity is far from complete and provides a basis to reveal largely unresolved mechanisms that coordinate transcription initiation and mRNA processing.
Conflict of interest statement
Ethics approval and consent to participate
No ethical approval was needed to perform this study.
Competing interests
ET and SWT are full-time employees of Pacific Biosciences. RHY and HEJ are full-time employees of LGC Biosearch Technologies. All other authors declare that they have no competing interests.
Review history
This article is part of our transparent review trial, and as such the review history is available as Additional file 2.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
