

CaMKII (Ca2+/Calmodulin-Dependent Kinase II) in Mitochondria of Smooth Muscle Cells Controls Mitochondrial Mobility, Migration, and Neointima Formation
- PMID: 29599132
- PMCID: PMC5970052
- DOI: 10.1161/ATVBAHA.118.310951
CaMKII (Ca2+/Calmodulin-Dependent Kinase II) in Mitochondria of Smooth Muscle Cells Controls Mitochondrial Mobility, Migration, and Neointima Formation
Abstract
Objective: The main objective of this study is to define the mechanisms by which mitochondria control vascular smooth muscle cell (VSMC) migration and impact neointimal hyperplasia.
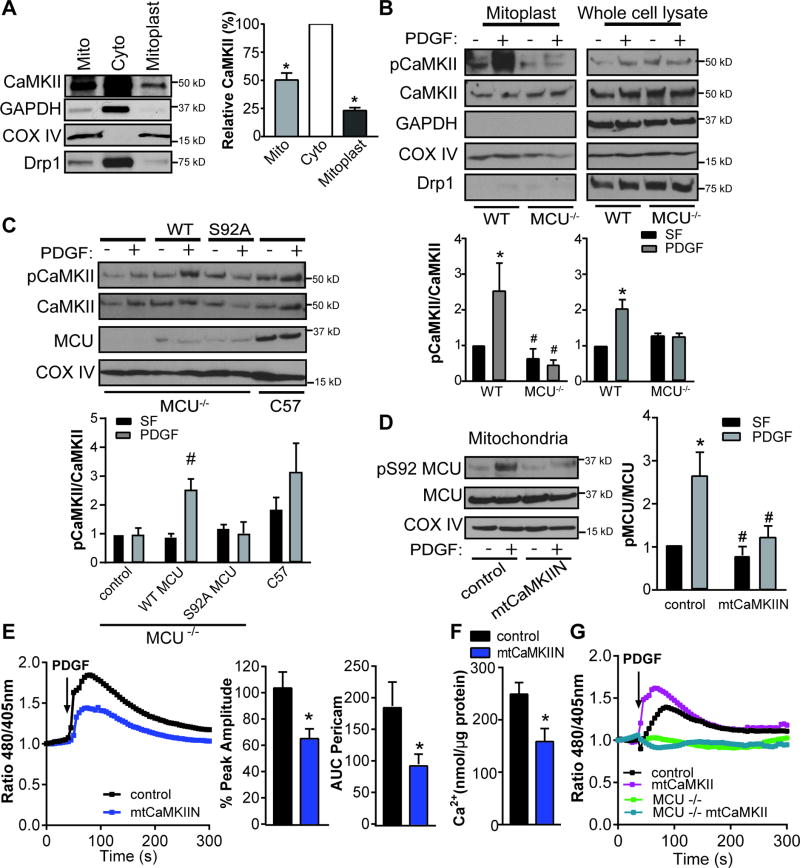
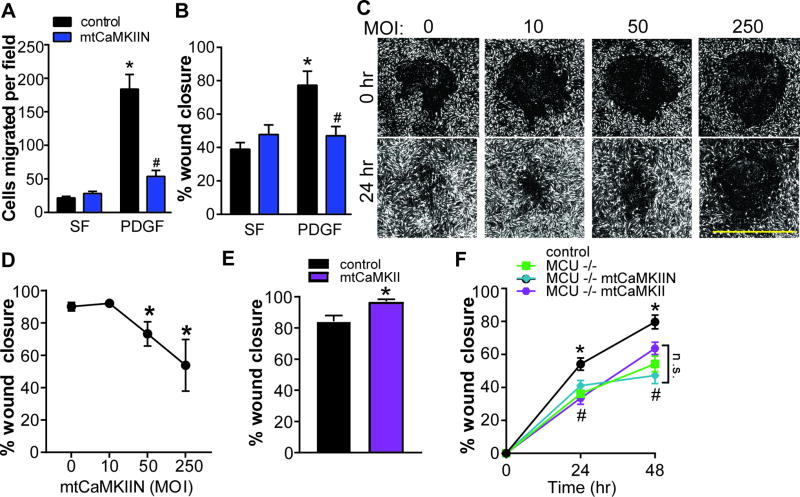
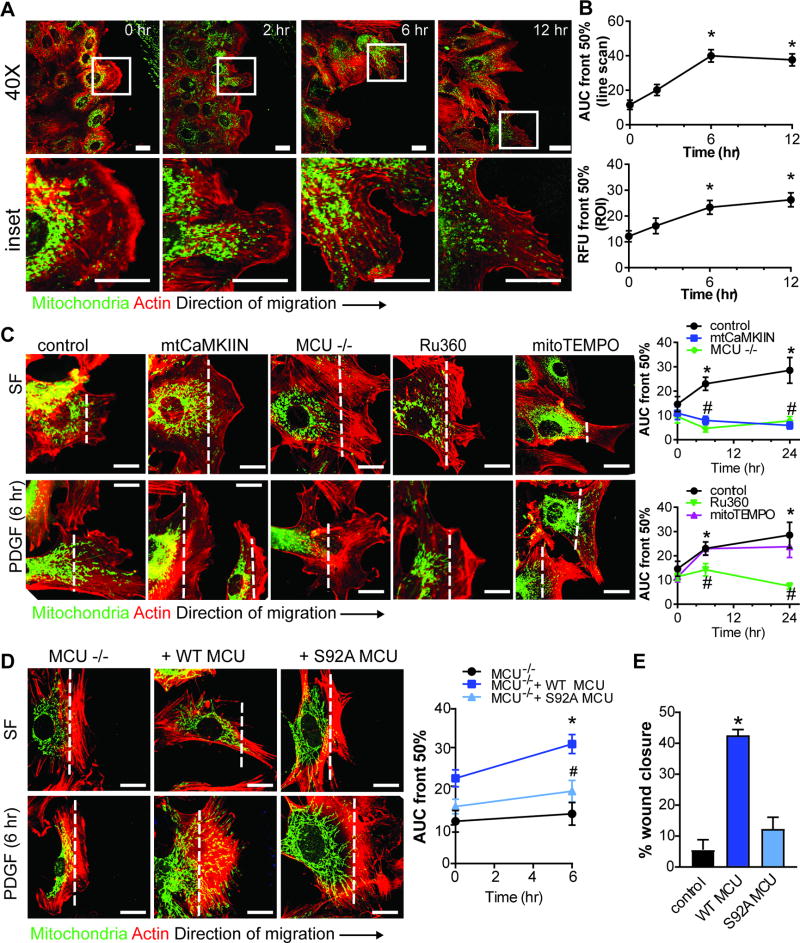
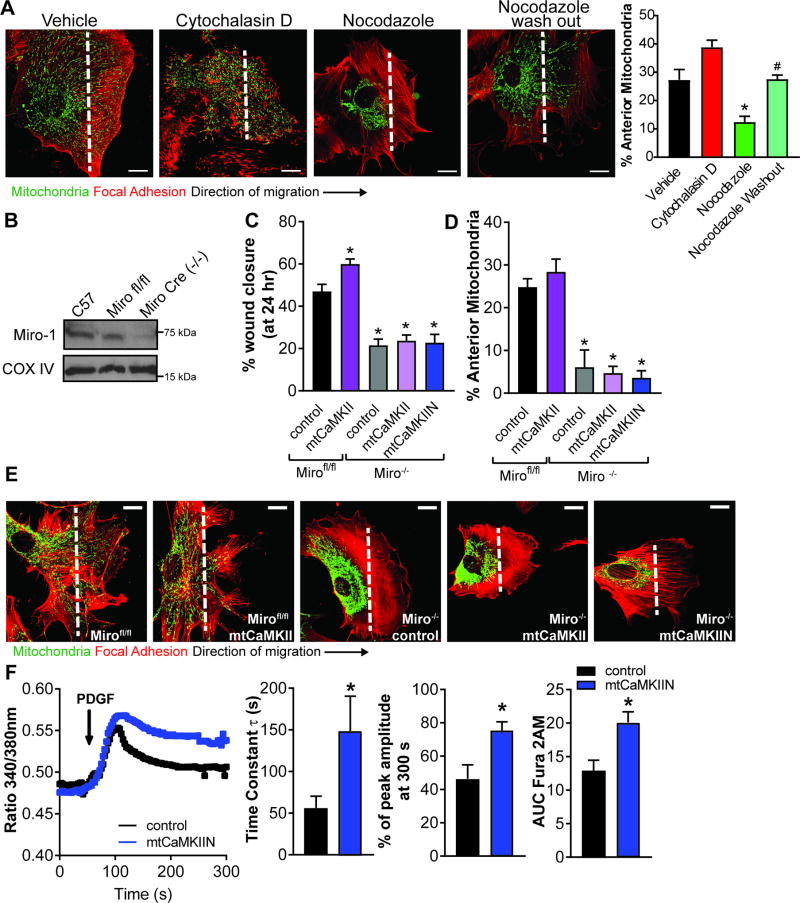
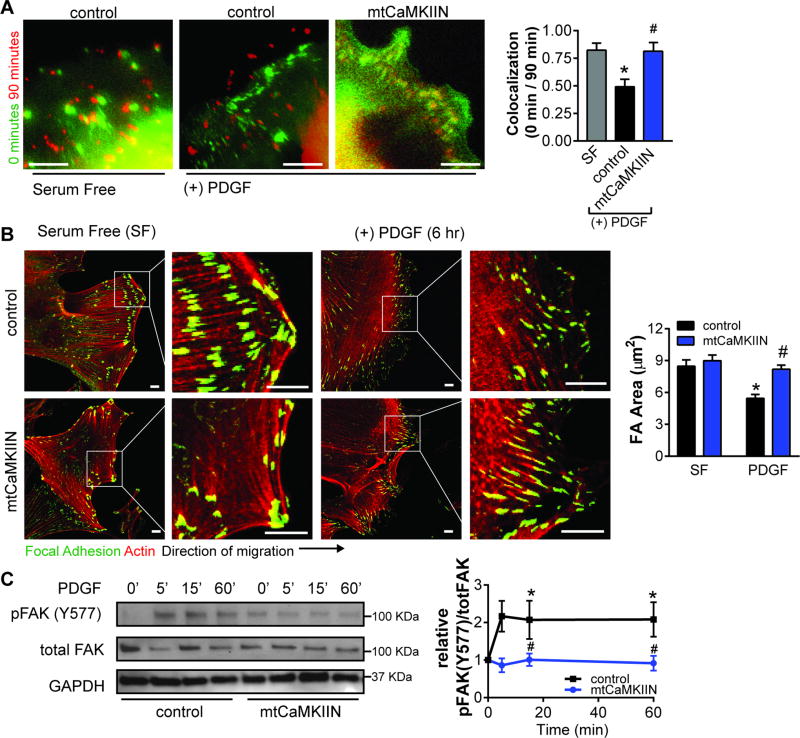
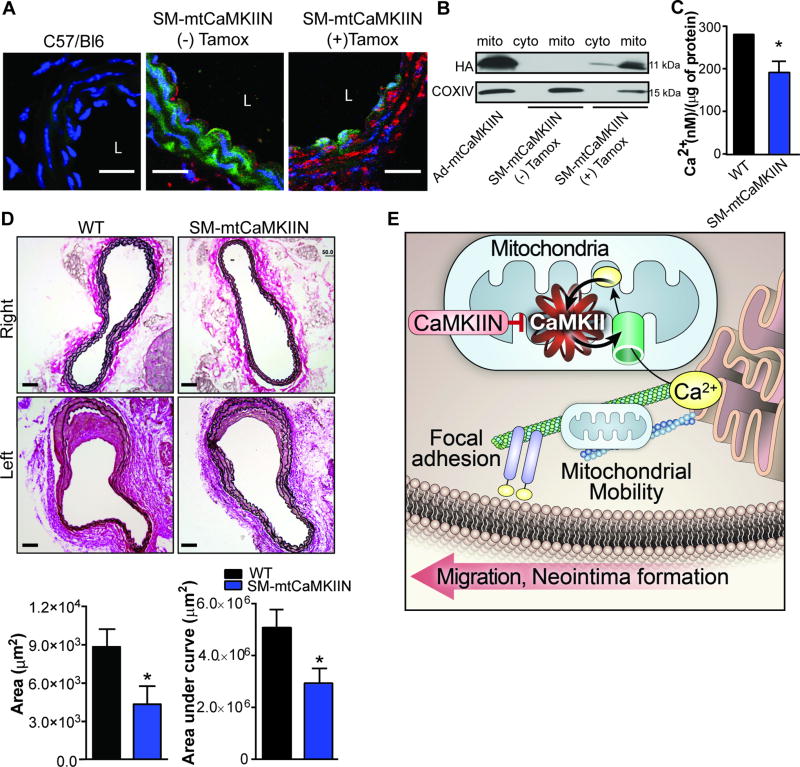
Approach and results: The multifunctional CaMKII (Ca2+/calmodulin-dependent kinase II) in the mitochondrial matrix of VSMC drove a feed-forward circuit with the mitochondrial Ca2+ uniporter (MCU) to promote matrix Ca2+ influx. MCU was necessary for the activation of mitochondrial CaMKII (mtCaMKII), whereas mtCaMKII phosphorylated MCU at the regulatory site S92 that promotes Ca2+ entry. mtCaMKII was necessary and sufficient for platelet-derived growth factor-induced mitochondrial Ca2+ uptake. This effect was dependent on MCU. mtCaMKII and MCU inhibition abrogated VSMC migration and mitochondrial translocation to the leading edge. Overexpression of wild-type MCU, but not MCU S92A, mutant in MCU-/- VSMC rescued migration and mitochondrial mobility. Inhibition of microtubule, but not of actin assembly, blocked mitochondrial mobility. The outer mitochondrial membrane GTPase Miro-1 promotes mitochondrial mobility via microtubule transport but arrests it in subcellular domains of high Ca2+ concentrations. In Miro-1-/- VSMC, mitochondrial mobility and VSMC migration were abolished, and overexpression of mtCaMKII or a CaMKII inhibitory peptide in mitochondria (mtCaMKIIN) had no effect. Consistently, inhibition of mtCaMKII increased and prolonged cytosolic Ca2+ transients. mtCaMKII inhibition diminished phosphorylation of focal adhesion kinase and myosin light chain, leading to reduced focal adhesion turnover and cytoskeletal remodeling. In a transgenic model of selective mitochondrial CaMKII inhibition in VSMC, neointimal hyperplasia was significantly reduced after vascular injury.
Conclusions: These findings identify mitochondrial CaMKII as a key regulator of mitochondrial Ca2+ uptake via MCU, thereby controlling mitochondrial translocation and VSMC migration after vascular injury.
Keywords: calcium; calcium-calmodulin-dependent protein kinase type 2; hyperplasia; microtubules; mitochondria; neointima.
© 2018 American Heart Association, Inc.
Conflict of interest statement
Figures
Comment in
-
Smooth Muscle Cells Move With Mitochondria.Arterioscler Thromb Vasc Biol. 2018 Jun;38(6):1255-1257. doi: 10.1161/ATVBAHA.118.311085. Arterioscler Thromb Vasc Biol. 2018. PMID: 29793991 Free PMC article. No abstract available.
Similar articles
-
Mitochondrial Calcium Uniporter Regulates Metabolic Remodeling and Smooth Muscle Cell Proliferation in Type 2 Diabetes.J Am Heart Assoc. 2025 Aug 5;14(15):e039220. doi: 10.1161/JAHA.124.039220. Epub 2025 Jul 17. J Am Heart Assoc. 2025. PMID: 40673552
-
Mixed-lineage kinase 3 deficiency promotes neointima formation through increased activation of the RhoA pathway in vascular smooth muscle cells.Arterioscler Thromb Vasc Biol. 2014 Jul;34(7):1429-36. doi: 10.1161/ATVBAHA.114.303439. Epub 2014 May 1. Arterioscler Thromb Vasc Biol. 2014. PMID: 24790140 Free PMC article.
-
Inositol 1,4,5-Trisphosphate Receptors Regulate Vascular Smooth Muscle Cell Proliferation and Neointima Formation in Mice.J Am Heart Assoc. 2024 Aug 6;13(15):e034203. doi: 10.1161/JAHA.124.034203. Epub 2024 Jul 18. J Am Heart Assoc. 2024. PMID: 39023067 Free PMC article.
-
The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease.Pflugers Arch. 2008 Aug;456(5):769-85. doi: 10.1007/s00424-008-0491-8. Epub 2008 Mar 26. Pflugers Arch. 2008. PMID: 18365243 Free PMC article. Review.
-
New insights into the role of mitochondrial calcium homeostasis in cell migration.Biochem Biophys Res Commun. 2018 May 27;500(1):75-86. doi: 10.1016/j.bbrc.2017.05.039. Epub 2017 May 8. Biochem Biophys Res Commun. 2018. PMID: 28495532 Free PMC article. Review.
Cited by
-
Mitochondrial CaMKII causes adverse metabolic reprogramming and dilated cardiomyopathy.Nat Commun. 2020 Sep 4;11(1):4416. doi: 10.1038/s41467-020-18165-6. Nat Commun. 2020. PMID: 32887881 Free PMC article.
-
Inhibition of CaMKII in mitochondria preserves endothelial barrier function after irradiation.Free Radic Biol Med. 2020 Jan;146:287-298. doi: 10.1016/j.freeradbiomed.2019.11.012. Epub 2019 Nov 9. Free Radic Biol Med. 2020. Retraction in: Free Radic Biol Med. 2022 Jul;187:204. doi: 10.1016/j.freeradbiomed.2022.05.021. PMID: 31711984 Free PMC article. Retracted.
-
The mitochondrial regulation of smooth muscle cell proliferation in type 2 diabetes.bioRxiv [Preprint]. 2023 Feb 16:2023.02.15.528765. doi: 10.1101/2023.02.15.528765. bioRxiv. 2023. Update in: Int J Mol Sci. 2023 Aug 17;24(16):12897. doi: 10.3390/ijms241612897. PMID: 36824758 Free PMC article. Updated. Preprint.
-
Metabolic Remodeling and Implicated Calcium and Signal Transduction Pathways in the Pathogenesis of Heart Failure.Int J Mol Sci. 2021 Sep 30;22(19):10579. doi: 10.3390/ijms221910579. Int J Mol Sci. 2021. PMID: 34638917 Free PMC article. Review.
-
Smooth Muscle Cells Move With Mitochondria.Arterioscler Thromb Vasc Biol. 2018 Jun;38(6):1255-1257. doi: 10.1161/ATVBAHA.118.311085. Arterioscler Thromb Vasc Biol. 2018. PMID: 29793991 Free PMC article. No abstract available.
References
-
- Moses JW, Leon MB, Popma JJ, Fitzgerald PJ, Holmes DR, O'Shaughnessy C, Caputo RP, Kereiakes DJ, Williams DO, Teirstein PS, Jaeger JL, Kuntz RE Investigators S. Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. The New England journal of medicine. 2003;349:1315–1323. - PubMed
-
- Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. 2007;100:460–473. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
