

Of Men and Mice: Modeling the Fragile X Syndrome
- PMID: 29599705
- PMCID: PMC5862809
- DOI: 10.3389/fnmol.2018.00041
Of Men and Mice: Modeling the Fragile X Syndrome
Abstract
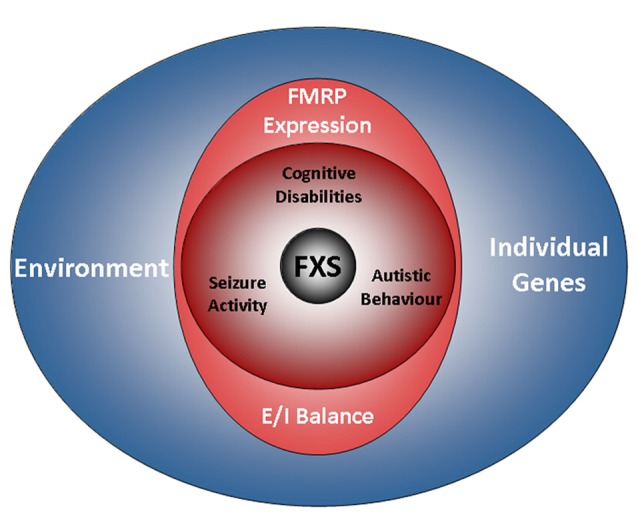
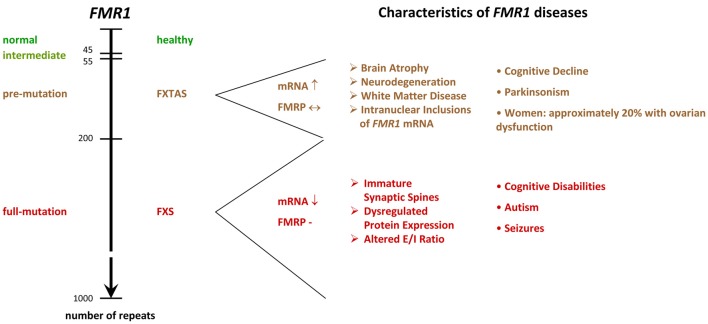
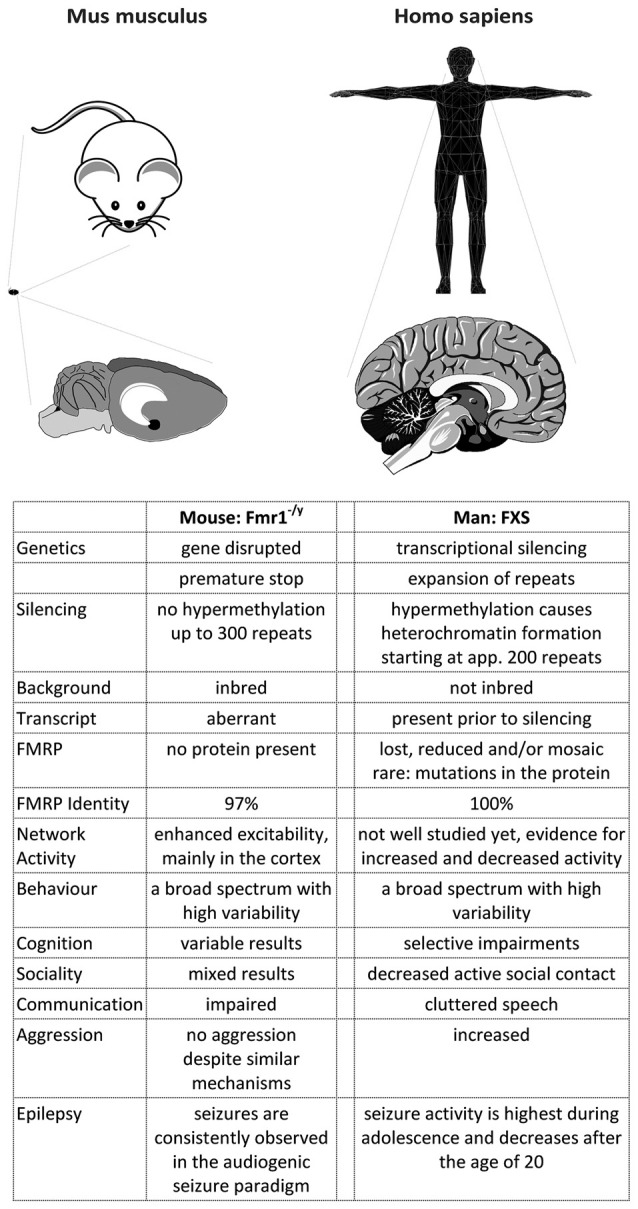
The Fragile X Syndrome (FXS) is one of the most common forms of inherited intellectual disability in all human societies. Caused by the transcriptional silencing of a single gene, the fragile x mental retardation gene FMR1, FXS is characterized by a variety of symptoms, which range from mental disabilities to autism and epilepsy. More than 20 years ago, a first animal model was described, the Fmr1 knock-out mouse. Several other models have been developed since then, including conditional knock-out mice, knock-out rats, a zebrafish and a drosophila model. Using these model systems, various targets for potential pharmaceutical treatments have been identified and many treatments have been shown to be efficient in preclinical studies. However, all attempts to turn these findings into a therapy for patients have failed thus far. In this review, I will discuss underlying difficulties and address potential alternatives for our future research.
Keywords: E/I balance; FMR1; Fragile X Syndrome; autism spectrum disorders; behavior and cognition; microsatellite instability; mouse model; primates.
Figures
References
-
- Ackerman S. (1992). Discovering the Brain. Washington, DC: National Academies Press. - PubMed
-
- Adihe Lokanga R., Zhao X.-N., Entezam A., Usdin K. (2014). X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: implications for the mechanism of repeat expansion. Hum. Mol. Genet. 23, 4985–4994. 10.1093/hmg/ddu213 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases