

Emerging Players at the Intersection of Chondrocyte Loss of Maturational Arrest, Oxidative Stress, Senescence and Low-Grade Inflammation in Osteoarthritis
- PMID: 29599894
- PMCID: PMC5828476
- DOI: 10.1155/2018/3075293
Emerging Players at the Intersection of Chondrocyte Loss of Maturational Arrest, Oxidative Stress, Senescence and Low-Grade Inflammation in Osteoarthritis
Abstract
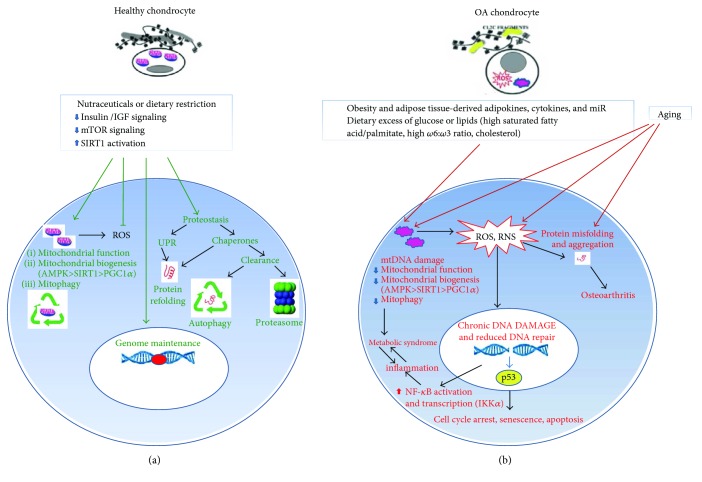
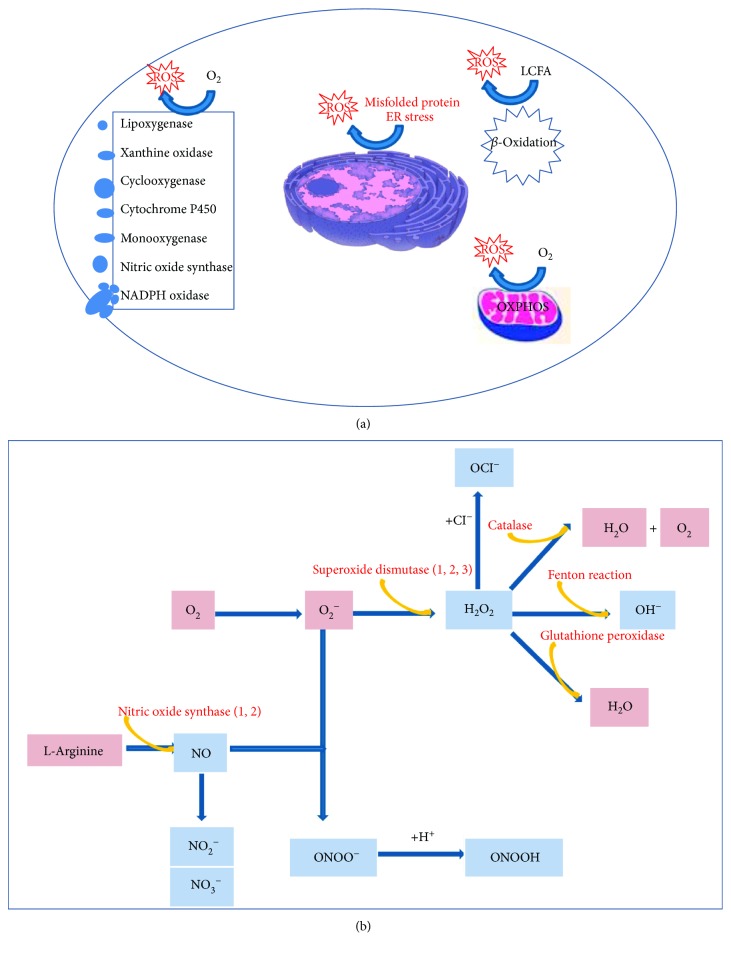
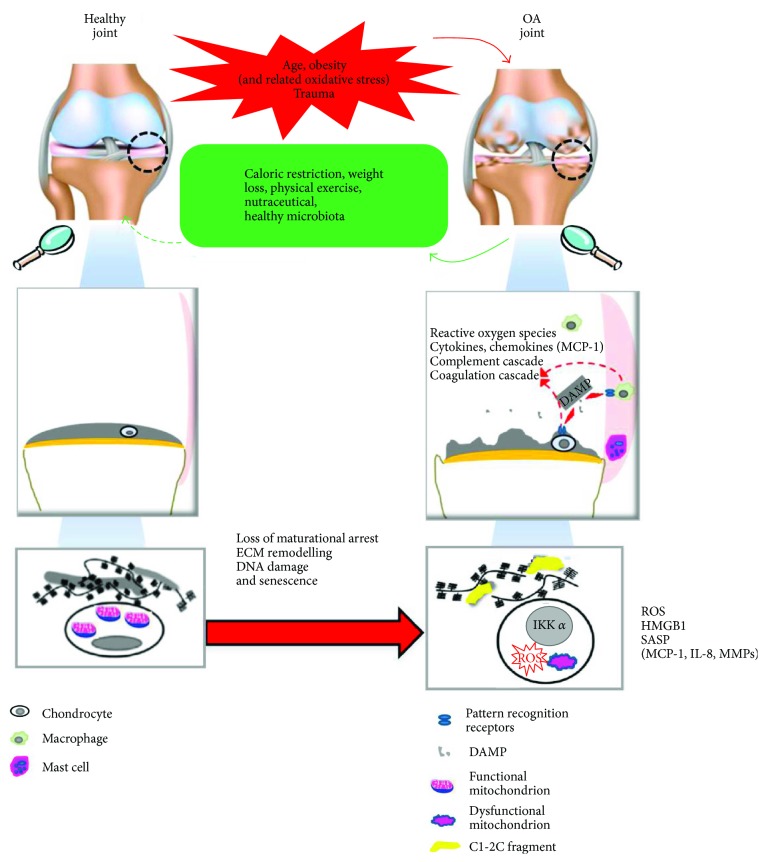
The prevalence of Osteoarthritis (OA) is increasing because of the progressive aging and unhealthy lifestyle. These risk factors trigger OA by removing constraints that keep the tightly regulated low turnover of the extracellular matrix (ECM) of articular cartilage, the correct chondrocyte phenotype, and the functionality of major homeostatic mechanisms, such as mitophagy, that allows for the clearance of dysfunctional mitochondria, preventing increased production of reactive oxygen species, oxidative stress, and senescence. After OA onset, the presence of ECM degradation products is perceived as a "danger" signal by the chondrocytes and the synovial macrophages that release alarmins with autocrine/paracrine effects on the same cells. Alarmins trigger innate immunity in the joint, with important systemic crosstalks that explain the beneficial effects of dietary interventions and improved lifestyle. Alarmins also boost low-grade inflammation: the release of inflammatory molecules and chemokines sustained by continuous triggering of NF-κB within an altered cellular setting that allows its higher transcriptional activity. Chemokines exert pleiotropic functions in OA, including the recruitment of inflammatory cells and the induction of ECM remodeling. Some chemokines have been successfully targeted to attenuate structural damage or pain in OA animal models. This represents a promising strategy for the future management of human OA.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
