

Intestinal Failure and Aberrant Lipid Metabolism in Patients With DGAT1 Deficiency
- PMID: 29604290
- PMCID: PMC6058035
- DOI: 10.1053/j.gastro.2018.03.040
Intestinal Failure and Aberrant Lipid Metabolism in Patients With DGAT1 Deficiency
Abstract
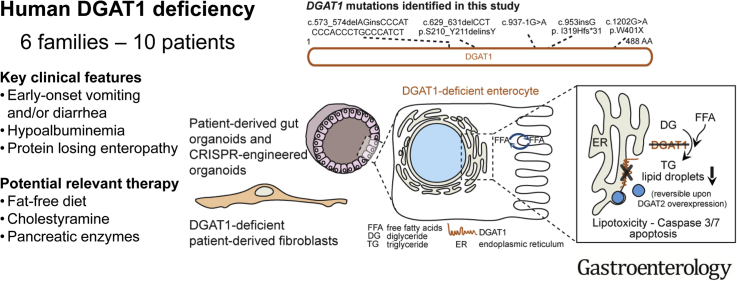
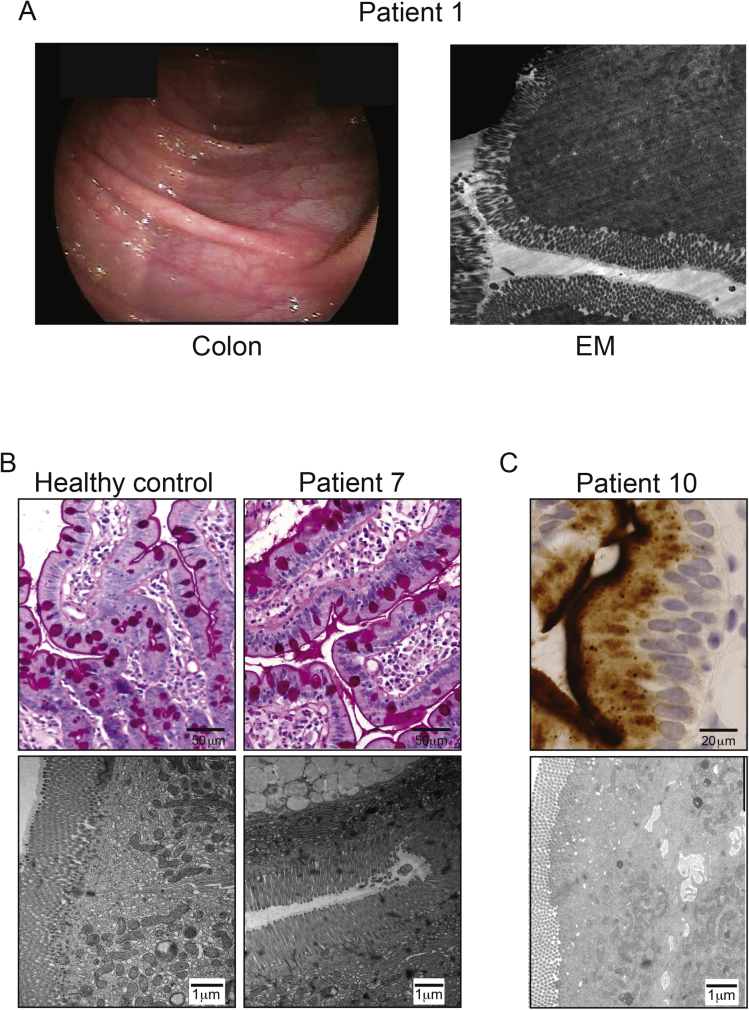
Background & aims: Congenital diarrheal disorders are rare inherited intestinal disorders characterized by intractable, sometimes life-threatening, diarrhea and nutrient malabsorption; some have been associated with mutations in diacylglycerol-acyltransferase 1 (DGAT1), which catalyzes formation of triacylglycerol from diacylglycerol and acyl-CoA. We investigated the mechanisms by which DGAT1 deficiency contributes to intestinal failure using patient-derived organoids.
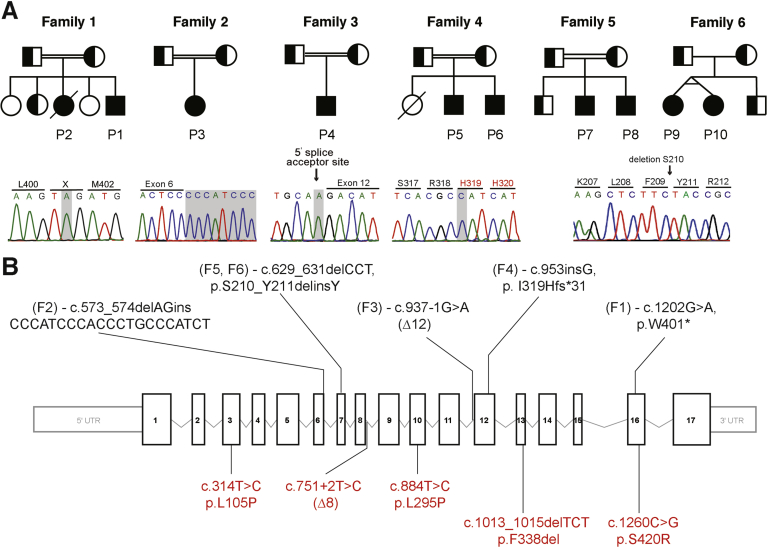
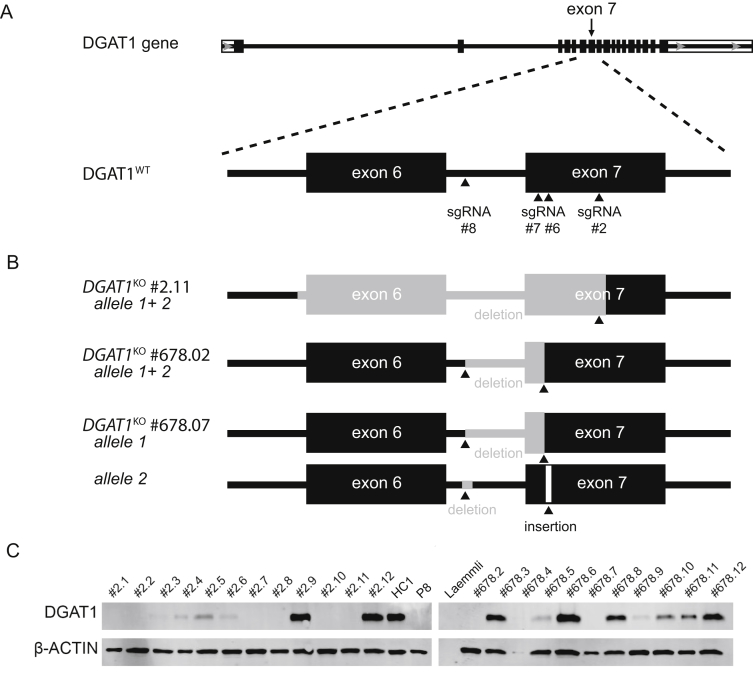
Methods: We collected blood samples from 10 patients, from 6 unrelated pedigrees, who presented with early-onset severe diarrhea and/or vomiting, hypoalbuminemia, and/or (fatal) protein-losing enteropathy with intestinal failure; we performed next-generation sequencing analysis of DNA from 8 patients. Organoids were generated from duodenal biopsies from 3 patients and 3 healthy individuals (controls). Caco-2 cells and patient-derived dermal fibroblasts were transfected or transduced with vectors that express full-length or mutant forms of DGAT1 or full-length DGAT2. We performed CRISPR/Cas9-guided disruption of DGAT1 in control intestinal organoids. Cells and organoids were analyzed by immunoblot, immunofluorescence, flow cytometry, chromatography, quantitative real-time polymerase chain reaction, and for the activity of caspases 3 and 7.
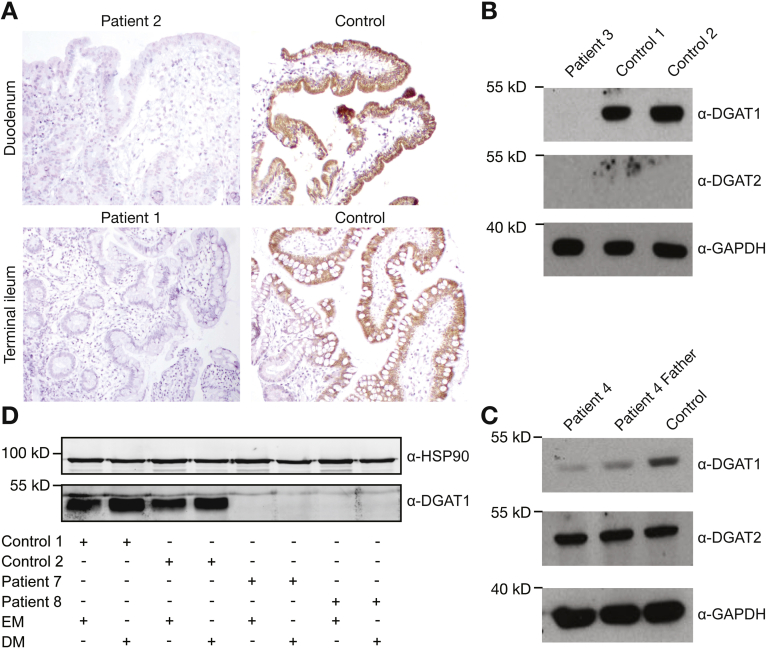
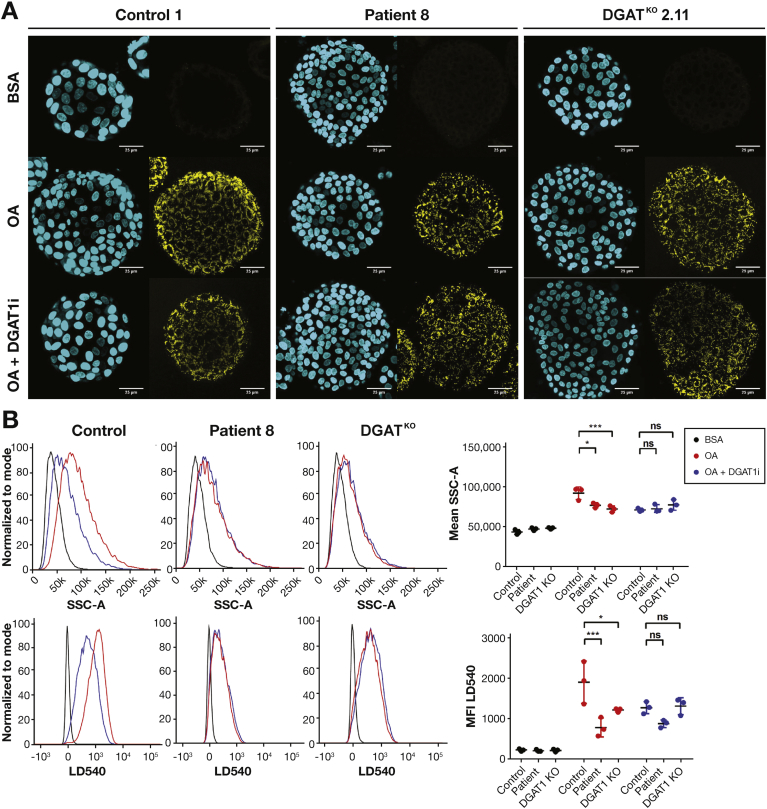
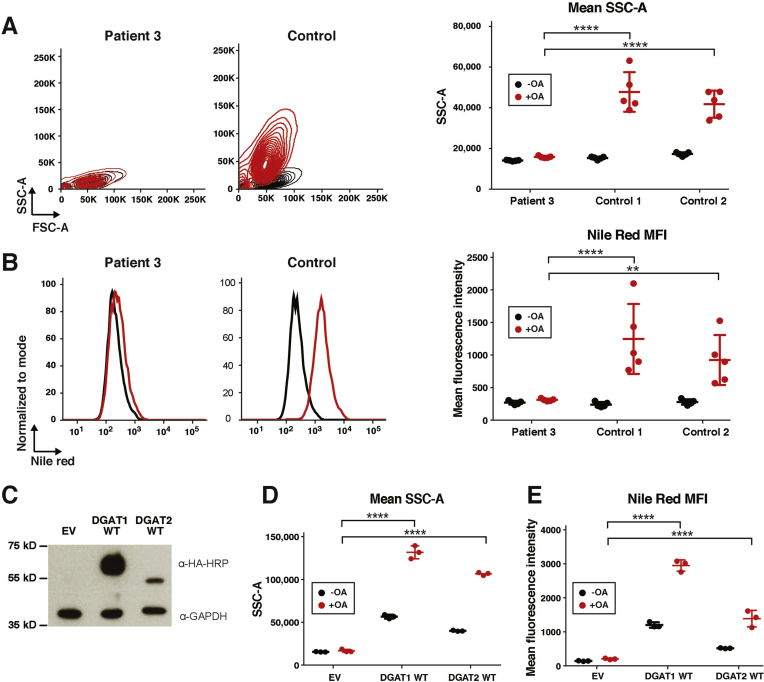
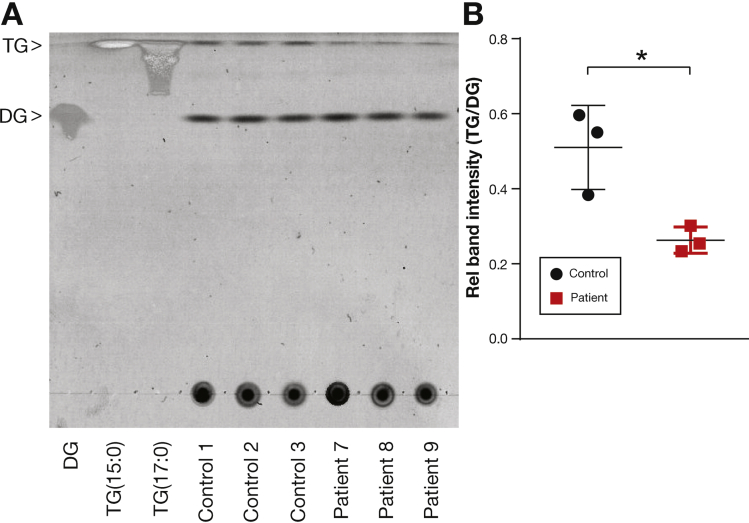
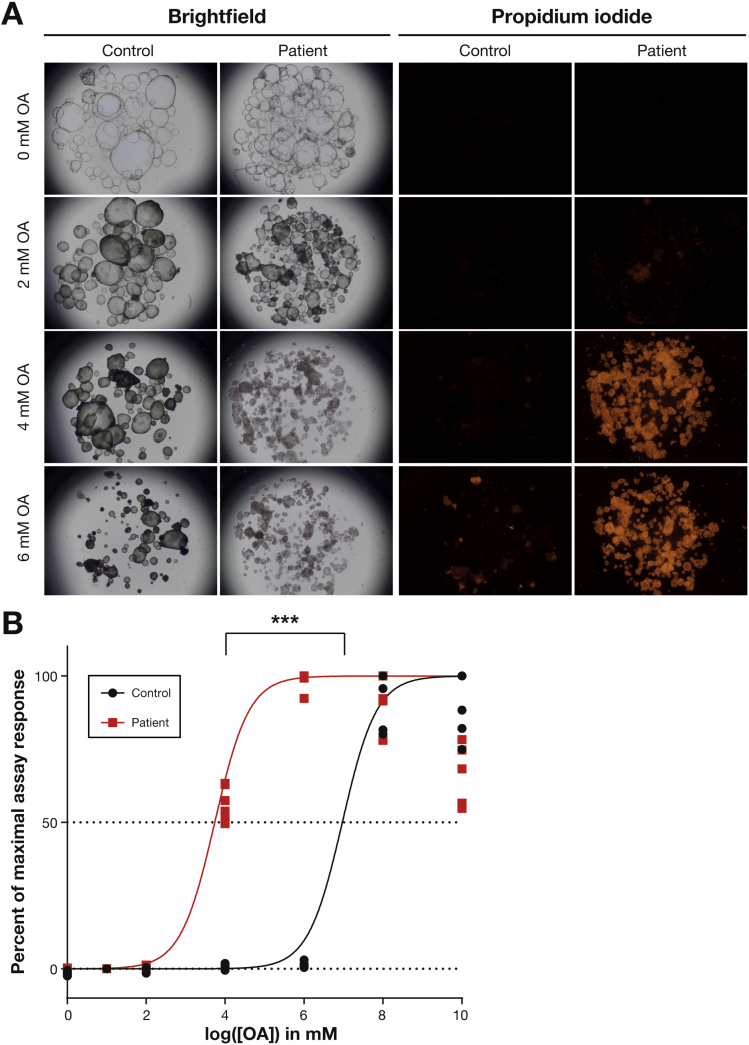
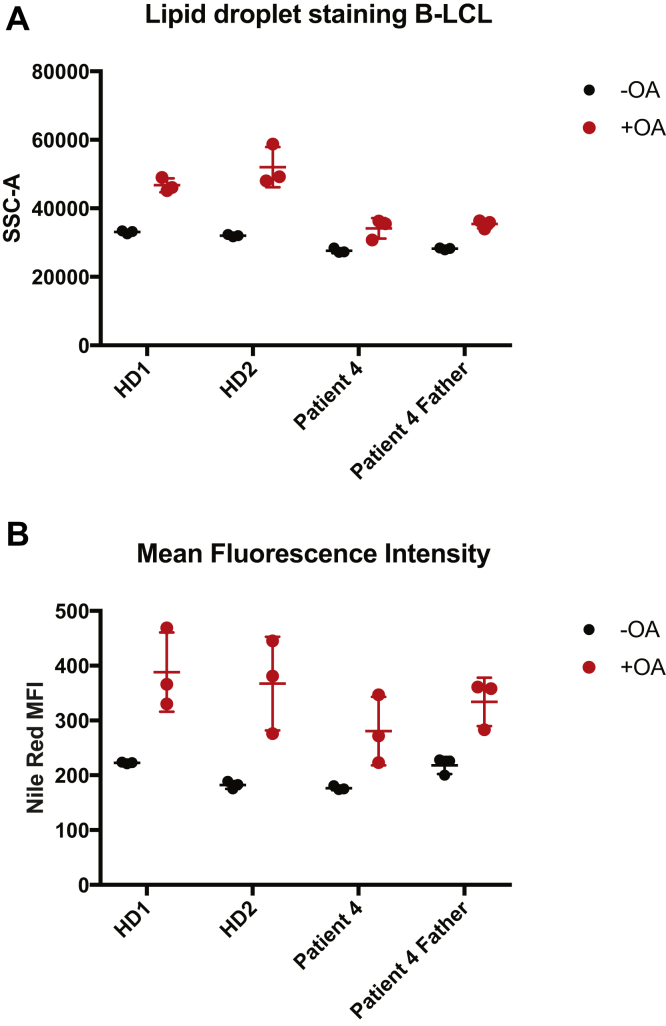
Results: In the 10 patients, we identified 5 bi-allelic loss-of-function mutations in DGAT1. In patient-derived fibroblasts and organoids, the mutations reduced expression of DGAT1 protein and altered triacylglycerol metabolism, resulting in decreased lipid droplet formation after oleic acid addition. Expression of full-length DGAT2 in patient-derived fibroblasts restored formation of lipid droplets. Organoids derived from patients with DGAT1 mutations were more susceptible to lipid-induced cell death than control organoids.
Conclusions: We identified a large cohort of patients with congenital diarrheal disorders with mutations in DGAT1 that reduced expression of its product; dermal fibroblasts and intestinal organoids derived from these patients had altered lipid metabolism and were susceptible to lipid-induced cell death. Expression of full-length wildtype DGAT1 or DGAT2 restored normal lipid metabolism in these cells. These findings indicate the importance of DGAT1 in fat metabolism and lipotoxicity in the intestinal epithelium. A fat-free diet might serve as the first line of therapy for patients with reduced DGAT1 expression. It is important to identify genetic variants associated with congenital diarrheal disorders for proper diagnosis and selection of treatment strategies.
Keywords: 3-D Culture Model; CDD; Genomic; PLE.
Copyright © 2018 AGA Institute. Published by Elsevier Inc. All rights reserved.
Figures
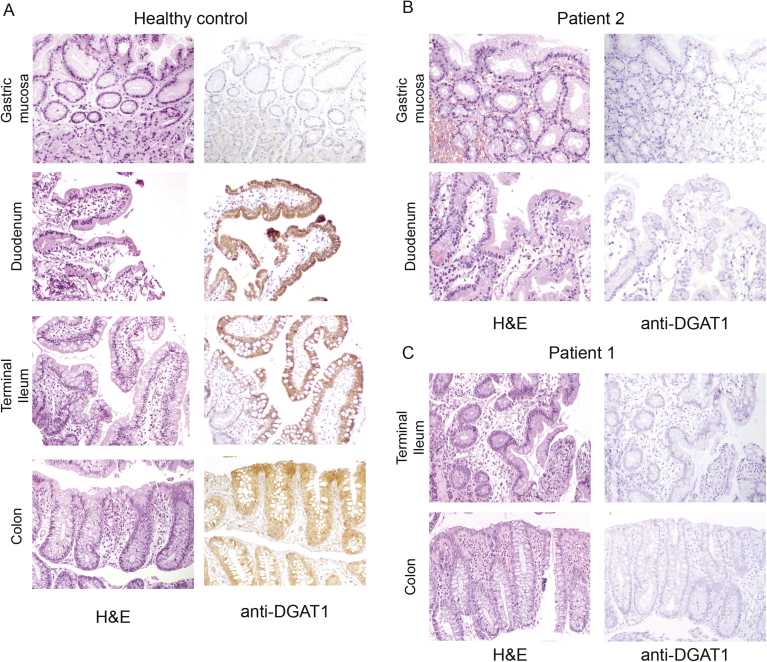
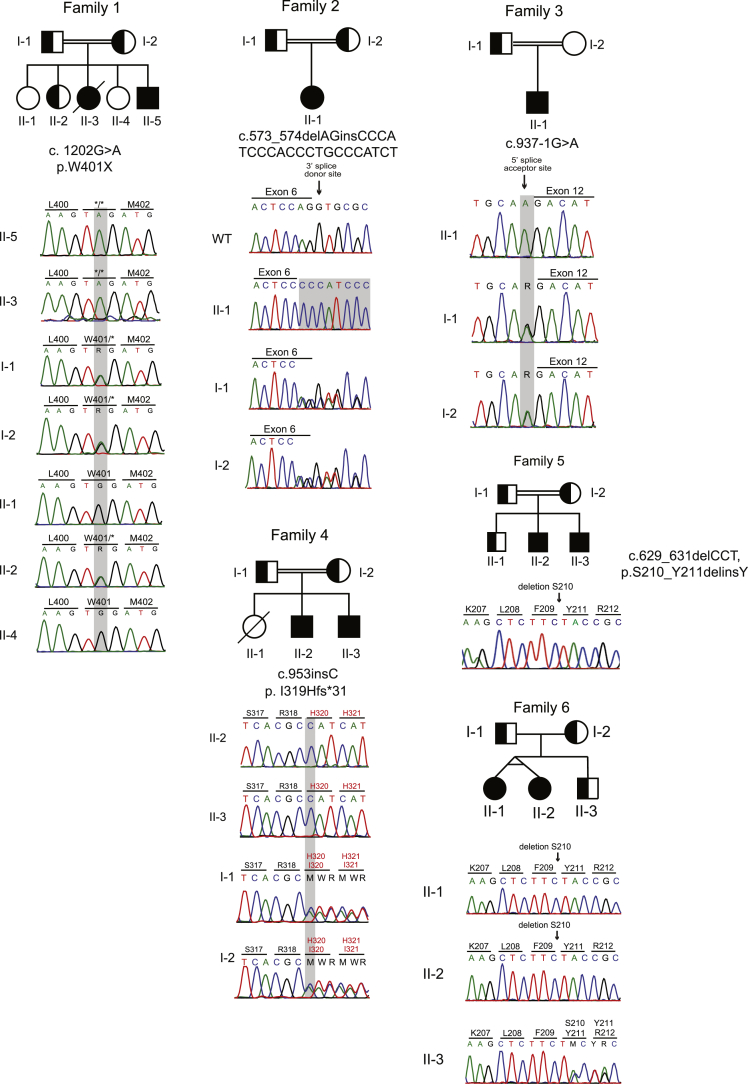
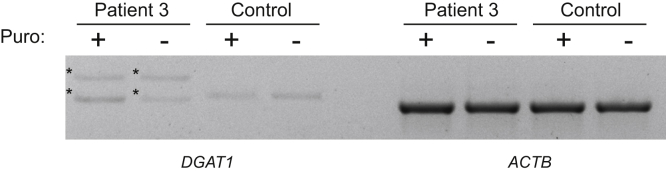
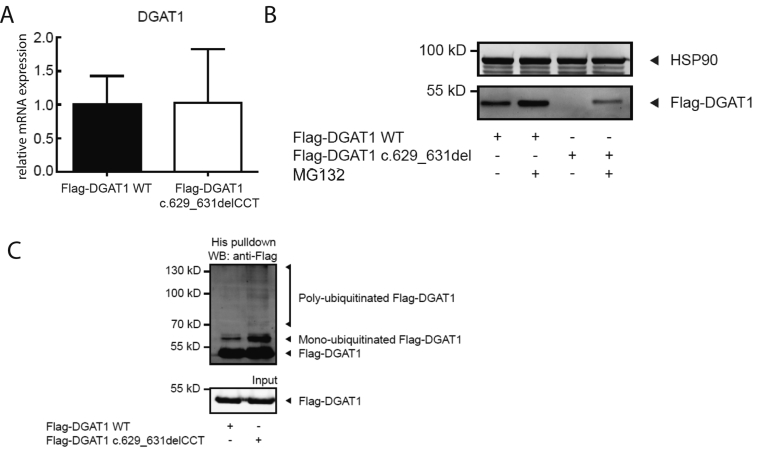
References
Supplementary References
-
- Wiegerinck C.L., Janecke A.R., Schneeberger K. Loss of syntaxin 3 causes variant microvillus inclusion disease. Gastroenterology. 2014;147:65–68.e10. - PubMed
-
- Fujii M., Matano M., Nanki K. Efficient genetic engineering of human intestinal organoids using electroporation. Nat Protoc. 2015;10:1474–1485. - PubMed
-
- Folch J., Lees M., Sloane Stanley G.H. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
