

Concomitant BCORL1 and BRAF Mutations in Vemurafenib-Resistant Melanoma Cells
- PMID: 29605720
- PMCID: PMC5915992
- DOI: 10.1016/j.neo.2018.02.009
Concomitant BCORL1 and BRAF Mutations in Vemurafenib-Resistant Melanoma Cells
Abstract
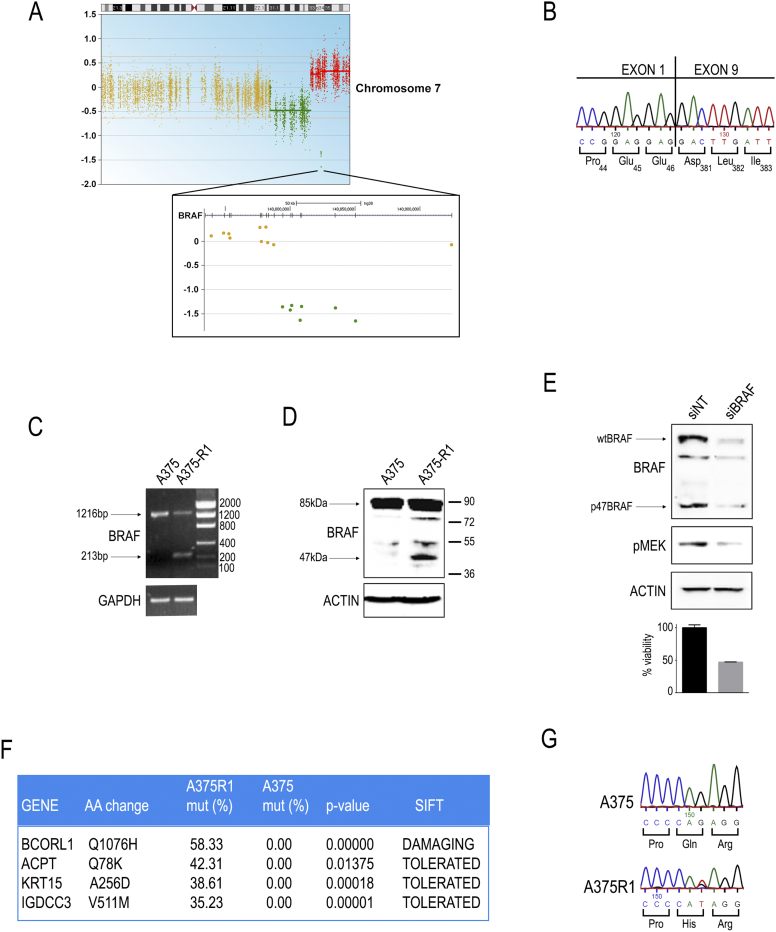
BRAF is the most frequently mutated gene in melanoma. Constitutive activation of mutant BRAFV600E leads to aberrant Ras-independent MAPK signaling and cell transformation. Inhibition of mutant BRAF is a current frontline therapy for such cases, with improved survival compared with chemotherapy. Unfortunately, reactivation of MAPK signaling by several mechanisms has been shown to cause drug resistance and disease recurrence. In this work, we describe the co-occurrence of an in-frame deletion within an amplified BRAFV600E locus and a missense point mutation of the transcriptional repressor BCORL1 in vemurafenib-resistant A375 melanoma cells. Functional data confirmed that truncated p47BRAFV600E and mutant BCORL1Q1076H both contribute to resistance. Interestingly, either endogenous BCORL1 silencing or ectopic BCORL1Q1076H expression mimicked the effects of a CRISPR/Cas9-edited BCORL1Q1076H locus, suggesting a complex mixture of loss- and gain-of-function effects caused by the mutation. Transcriptomic data confirmed this hypothesis. Finally, we show that the pan-RAF inhibitor sorafenib is not affected by expression of BRAF deletion variant and effectively synergizes with vemurafenib to block resistant cells, suggesting a possible intervention for this class of mutants.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures





References
-
- Gambacorti-Passerini C, Piazza R. How I treat newly diagnosed chronic myeloid leukemia in 2015. Am J Hematol. 2015;90(2):156–161. - PubMed
-
- Gambacorti Passerini C, Farina F, Stasia A, Redaelli S, Ceccon M, Mologni L, Messa C, Guerra L, Giudici G, Sala E. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J Natl Cancer Inst. 2014;106(2) - PubMed
-
- Krajewska J, Olczyk T, Jarzab B. Cabozantinib for the treatment of progressive metastatic medullary thyroid cancer. Expert Rev Clin Pharmacol. 2016;9(1):69–79. - PubMed
-
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
