

Modeling Structure and Dynamics of Protein Complexes with SAXS Profiles
- PMID: 29605933
- PMCID: PMC6022765
- DOI: 10.1007/978-1-4939-7759-8_29
Modeling Structure and Dynamics of Protein Complexes with SAXS Profiles
Abstract
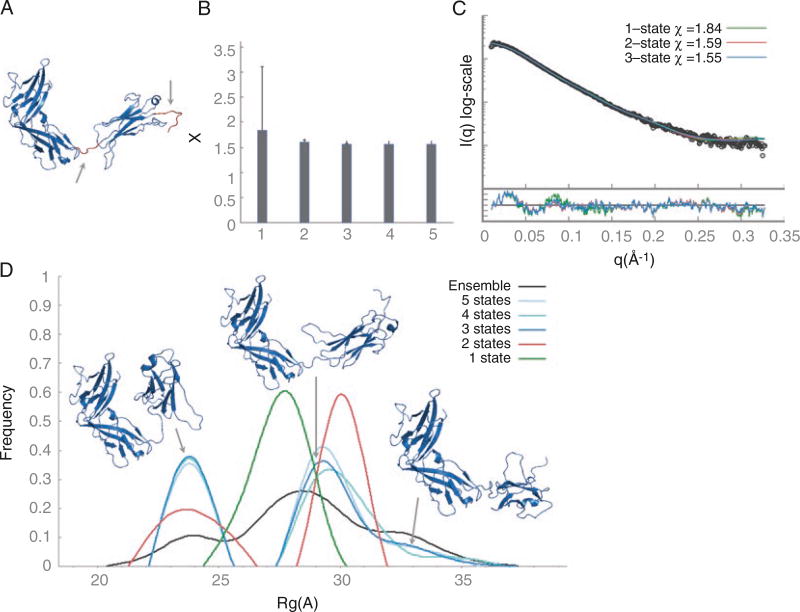
Small-angle X-ray scattering (SAXS) is an increasingly common and useful technique for structural characterization of molecules in solution. A SAXS experiment determines the scattering intensity of a molecule as a function of spatial frequency, termed SAXS profile. SAXS profiles can be utilized in a variety of molecular modeling applications, such as comparing solution and crystal structures, structural characterization of flexible proteins, assembly of multi-protein complexes, and modeling of missing regions in the high-resolution structure. Here, we describe protocols for modeling atomic structures based on SAXS profiles. The first protocol is for comparing solution and crystal structures including modeling of missing regions and determination of the oligomeric state. The second protocol performs multi-state modeling by finding a set of conformations and their weights that fit the SAXS profile starting from a single-input structure. The third protocol is for protein-protein docking based on the SAXS profile of the complex. We describe the underlying software, followed by demonstrating their application on interleukin 33 (IL33) with its primary receptor ST2 and DNA ligase IV-XRCC4 complex.
Keywords: Conformational ensembles; Conformational heterogeneity; Multi-state models; Protein-protein docking; Small-angle X-ray scattering (SAXS).
Figures





References
-
- Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang SJ, Scott JW, Dillard BD, Adams MW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6(8):606–612. https://doi.org/10.1038/nmeth.1353.nmeth.1353 [pii] - DOI - PMC - PubMed
-
- Hura GL, Budworth H, Dyer KN, Rambo RP, Hammel M, McMurray CT, Tainer JA. Comprehensive macromolecular conformations mapped by quantitative SAXS analyses. Nat Methods. 2013;10(6):453–454. https://doi.org/10.1038/nmeth.2453. - DOI - PMC - PubMed
-
- Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, Tainer JA, Hura GL. High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol Biol. 2014;1091:245–258. https://doi.org/10.1007/978-1-62703-691-7_18. - DOI - PMC - PubMed
-
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40(3):191–285. https://doi.org/10.1017/S0033583507004635. S0033583507004635 [pii] - DOI - PubMed
-
- Rambo RP, Tainer JA. Super-resolution in solution x-ray scattering and its applications to structural systems biology. Annu Rev Biophys. 2013;42:415–441. https://doi.org/10.1146/annurev-biophys-083012-130301. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
