

Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome
- PMID: 29606302
- PMCID: PMC5985336
- DOI: 10.1016/j.ajhg.2018.02.018
Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome
Abstract
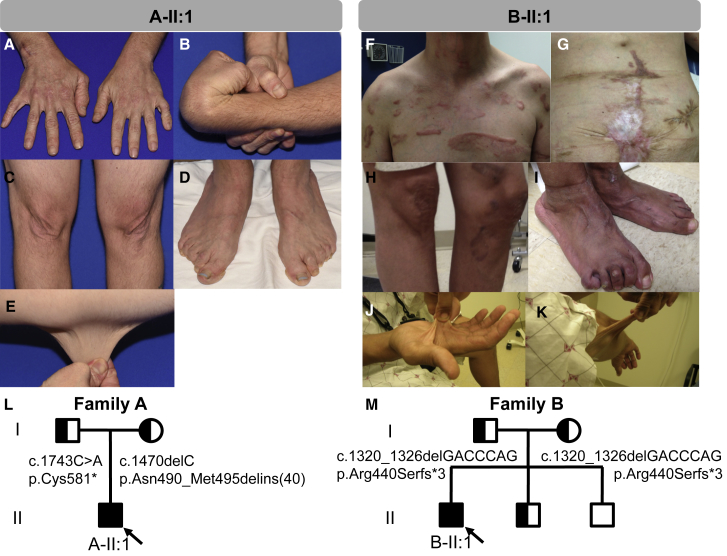
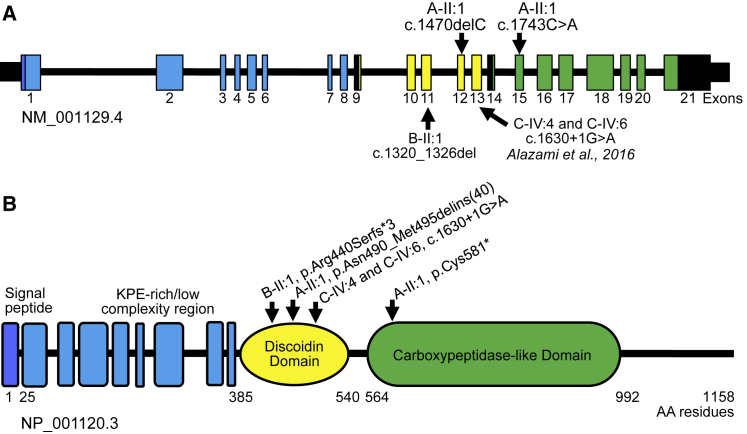
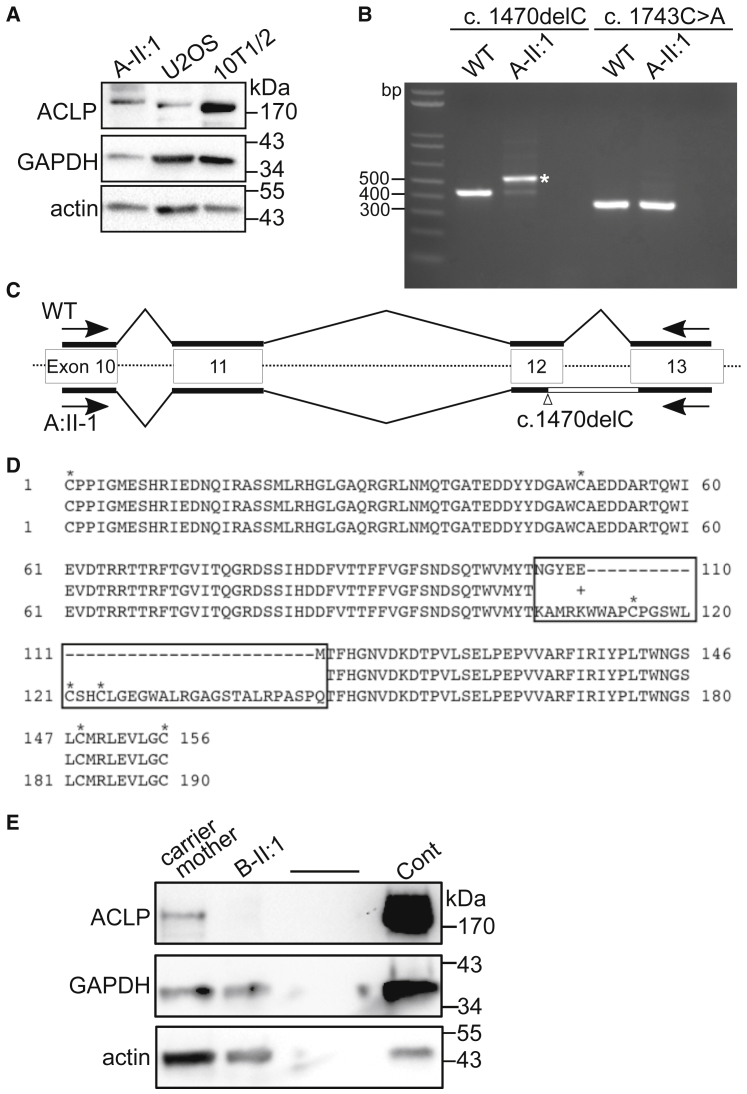
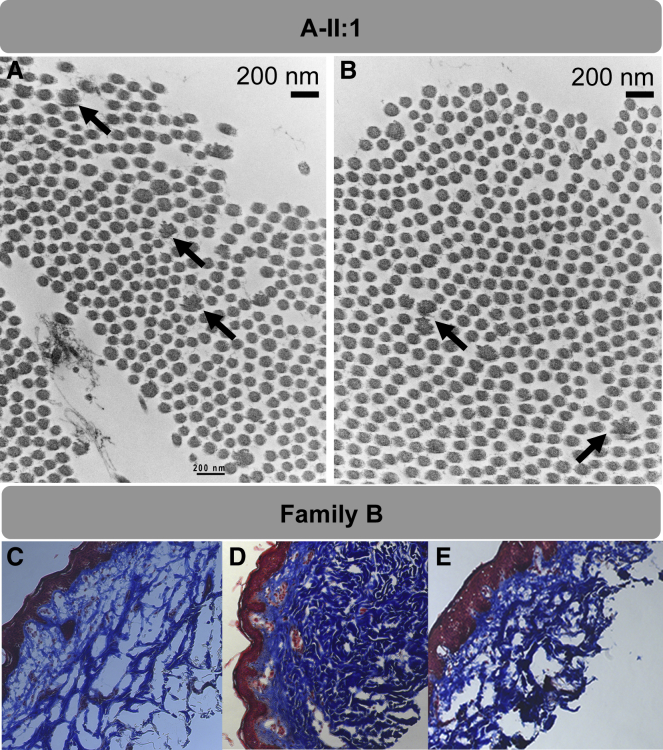
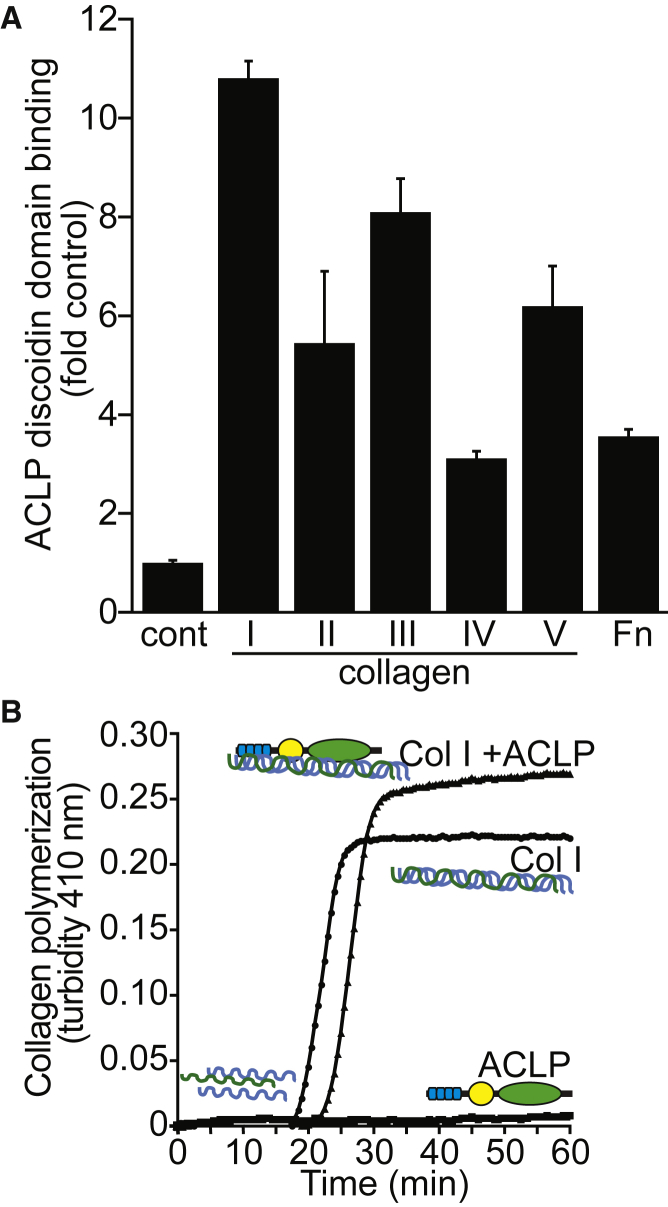
AEBP1 encodes the aortic carboxypeptidase-like protein (ACLP) that associates with collagens in the extracellular matrix (ECM) and has several roles in development, tissue repair, and fibrosis. ACLP is expressed in bone, the vasculature, and dermal tissues and is involved in fibroblast proliferation and mesenchymal stem cell differentiation into collagen-producing cells. Aebp1-/- mice have abnormal, delayed wound repair correlating with defects in fibroblast proliferation. In this study, we describe four individuals from three unrelated families that presented with a unique constellation of clinical findings including joint laxity, redundant and hyperextensible skin, poor wound healing with abnormal scarring, osteoporosis, and other features reminiscent of Ehlers-Danlos syndrome (EDS). Analysis of skin biopsies revealed decreased dermal collagen with abnormal collagen fibrils that were ragged in appearance. Exome sequencing revealed compound heterozygous variants in AEBP1 (c.1470delC [p.Asn490_Met495delins(40)] and c.1743C>A [p.Cys581∗]) in the first individual, a homozygous variant (c.1320_1326del [p.Arg440Serfs∗3]) in the second individual, and a homozygous splice site variant (c.1630+1G>A) in two siblings from the third family. We show that ACLP enhances collagen polymerization and binds to several fibrillar collagens via its discoidin domain. These studies support the conclusion that bi-allelic pathogenic variants in AEBP1 are the cause of this autosomal-recessive EDS subtype.
Keywords: ACLP; AEBP1; Aebp1-null mice; Ehlers-Danlos syndrome; aortic carboxypeptidase-like protein; collagen polymerization; connective tissue disorders; discoidin domain; exome sequencing; extracellular matrix.
Copyright © 2018 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Murphy-Ryan M., Psychogios A., Lindor N.M. Hereditary disorders of connective tissue: a guide to the emerging differential diagnosis. Genet. Med. 2010;12:344–354. - PubMed
-
- Vanakker O., Callewaert B., Malfait F., Coucke P. The genetics of soft connective tissue disorders. Annu. Rev. Genomics Hum. Genet. 2015;16:229–255. - PubMed
-
- Byers P.H., Murray M.L. Heritable collagen disorders: the paradigm of the Ehlers-Danlos syndrome. J. Invest. Dermatol. 2012;132:E6–E11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
