

Senescent cells: a therapeutic target for cardiovascular disease
- PMID: 29608141
- PMCID: PMC5873883
- DOI: 10.1172/JCI95146
Senescent cells: a therapeutic target for cardiovascular disease
Abstract
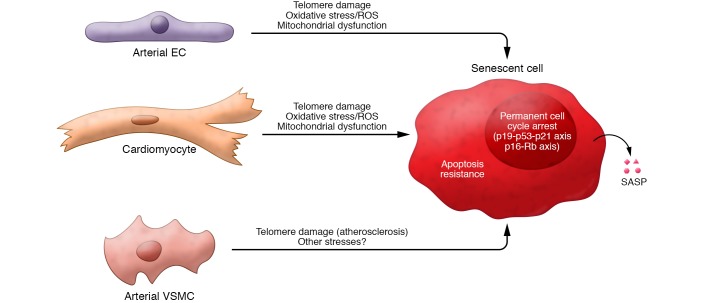
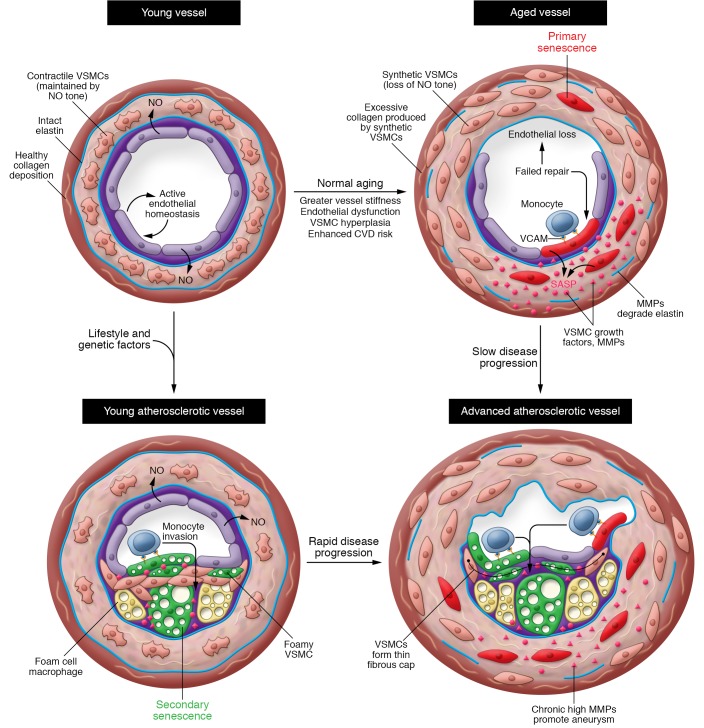
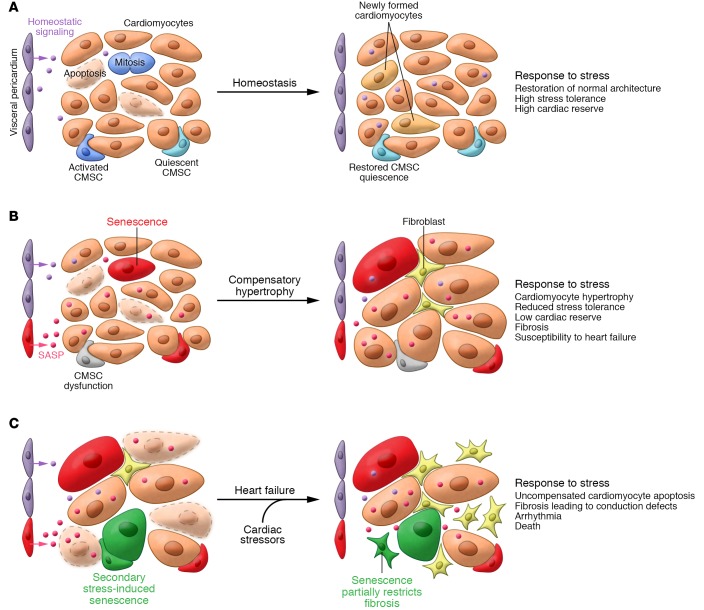
Cellular senescence, a major tumor-suppressive cell fate, has emerged from humble beginnings as an in vitro phenomenon into recognition as a fundamental mechanism of aging. In the process, senescent cells have attracted attention as a therapeutic target for age-related diseases, including cardiovascular disease (CVD), the leading cause of morbidity and mortality in the elderly. Given the aging global population and the inadequacy of current medical management, attenuating the health care burden of CVD would be transformative to clinical practice. Here, we review the evidence that cellular senescence drives CVD in a bimodal fashion by both priming the aged cardiovascular system for disease and driving established disease forward. Hence, the growing field of senotherapy (neutralizing senescent cells for therapeutic benefit) is poised to contribute to both prevention and treatment of CVD.
Conflict of interest statement
Figures
References
-
- Feingold KR, Grunfeld C. Approach to the patient with dyslipidemia. In: De Groot G, et al., eds. Endotext [Internet]. South Dartmouth, Massachusetts, USA; 2000. https://www.ncbi.nlm.nih.gov/books/NBK278943/ Accessed January 29, 2018.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
