

16p11.2 microdeletion syndrome: a case report
- PMID: 29609622
- PMCID: PMC5881179
- DOI: 10.1186/s13256-018-1587-1
16p11.2 microdeletion syndrome: a case report
Abstract
Background: The recurrent ∼ 600 kb 16p11.2 microdeletion is among the most commonly known genetic etiologies of autism spectrum disorder, overweightness, and related neurodevelopmental disorders.
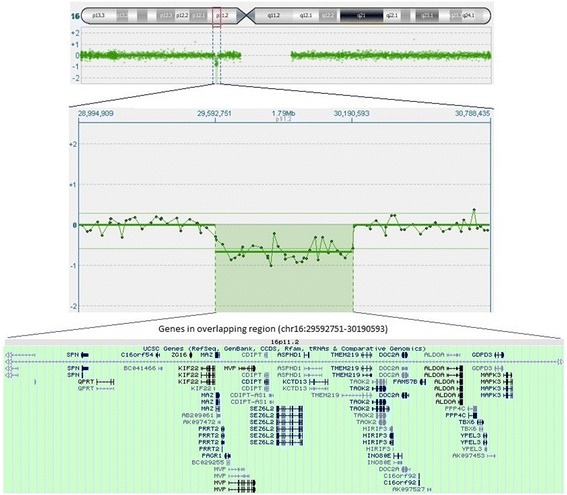
Case presentation: Our patient is a 2-year-old white girl from the first pregnancy of a non-consanguineous healthy young white couple (father 33-years old and mother 29-years old). Our patient and her parents' DNA were analyzed by comparative genomic hybridization-array platform. Comparative genomic hybridization-array analysis highlighted a ∼ 600 kb deletion in 16p11.2 region. It has a segregant nature, since it was found in the mother and in her 2-year-old daughter. The microdeletion was confirmed by fluorescence in situ hybridization analysis.
Conclusions: The presented clinical case is worthy of note since the observed microdeletion is often associated with a clinical phenotype tending to overweightness, but the proband (female) was hospitalized due to poor height and weight development, and anorexia. Moreover, the segregant nature of the observed genomic abnormality has to be noted, as well as the phenotypic variability between the mother and daughter. The case described here enriches the phenotypical spectrum linked to the 16p11.2 microdeletion. For these reasons, in the presence of a suspected genetic pathology it is fundamental to study the proband from the clinical point of view, to extend the clinical observation to the parents, and to provide a good family anamnesis. In this way, it is possible to reveal the presence of a familial genetic pathology whose phenotypical outcomes can be highly variable among the members of a family.
Keywords: 16p11.2 microdeletion syndrome; CGH-array; Developmental delay; Intellectual disability; Submicroscopic chromosomal changes.
Conflict of interest statement
Ethics approval and consent to participate
Ethical clearance was sought from the Directorate of Research of the Ethical Committee Basilicata Region (Italy).
Consent for publication
Written informed consent was obtained from the patient’s legal guardian(s) for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Jacquemont S, Reymond A, Zufferey F, Harewood L, Walters RG, Kutalik Z, Martinet D, Shen Y, Valsesia A, Beckmann ND, Thorleifsson G, Belfiore M, Bouquillon S, Campion D, de Leeuw N, de Vries BB, Esko T, Fernandez BA, Fernandez-Aranda F, Fernandez-Real JM, Gratacos M, Guilmatre A, Hoyer J, Jarvelin MR, Kooy RF, Kurg A, Le Caignec C, Mannik K, Platt OS, Sanlaville D, Van Haelst MM, Villatoro Gomez S, Walha F, Wu BL, Yu Y, Aboura A, Addor MC, Alembik Y, Antonarakis SE, Arveiler B, Barth M, Bednarek N, Bena F, Bergmann S, Beri M, Bernardini L, Blaumeiser B, Bonneau D, Bottani A, Boute O, Brunner HG, Cailley D, Callier P, Chiesa J, Chrast J, Coin L, Coutton C, Cuisset JM, Cuvellier JC, David A, de Freminville B, Delobel B, Delrue MA, Demeer B, Descamps D, Didelot G, Dieterich K, Disciglio V, Doco-Fenzy M, Drunat S, Duban-Bedu B, Dubourg C, El-Sayed Moustafa JS, Elliott P, Faas BH, Faivre L, Faudet A, Fellmann F, Ferrarini A, Fisher R, Flori E, Forer L, Gaillard D, Gerard M, Gieger C, Gimelli S, Gimelli G, Grabe HJ, Guichet A, Guillin O, Hartikainen AL, Heron D, Hippolyte L, Holder M, Homuth G, Isidor B, Jaillard S, Jaros Z, Jimenez-Murcia S, Helas GJ, Jonveaux P, Kaksonen S, Keren B, Kloss-Brandstatter A, Knoers NV, Koolen DA, Kroisel PM, Kronenberg F, Labalme A, Landais E, Lapi E, Layet V, Legallic S, Leheup B, Leube B, Lewis S, Lucas J, KD MD, Magnusson P, Marshall C, Mathieu-Dramard M, MI MC, Meitinger T, Mencarelli MA, Merla G, Moerman A, Mooser V, Morice-Picard F, Mucciolo M, Nauck M, Ndiaye NC, Nordgren A, Pasquier L, Petit F, Pfundt R, Plessis G, Rajcan-Separovic E, Ramelli GP, Rauch A, Ravazzolo R, Reis A, Renieri A, Richart C, Ried JS, Rieubland C, Roberts W, Roetzer KM, Rooryck C, Rossi M, Saemundsen E, Satre V, Schurmann C, Sigurdsson E, Stavropoulos DJ, Stefansson H, Tengstrom C, Thorsteinsdottir U, Tinahones FJ, Touraine R, Vallee L, van Binsbergen E, Van der Aa N, Vincent-Delorme C, Visvikis-Siest S, Vollenweider P, Volzke H, Vulto-van Silfhout AT, Waeber G, Wallgren-Pettersson C, Witwicki RM, Zwolinksi S, Andrieux J, Estivill X, Gusella JF, Gustafsson O, Metspalu A, Scherer SW, Stefansson K, Blakemore AI, Beckmann JS, Froguel P. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature. 2011;478:97–102. doi: 10.1038/nature10406. - DOI - PMC - PubMed
-
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R, Zwaigenbaum L, Roberts W, Fernandez B, Szatmari P, Scherer SW. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. - DOI - PMC - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
