

Evolutionary Origins of Enteric Hepatitis Viruses
- PMID: 29610146
- PMCID: PMC6280709
- DOI: 10.1101/cshperspect.a031690
Evolutionary Origins of Enteric Hepatitis Viruses
Abstract
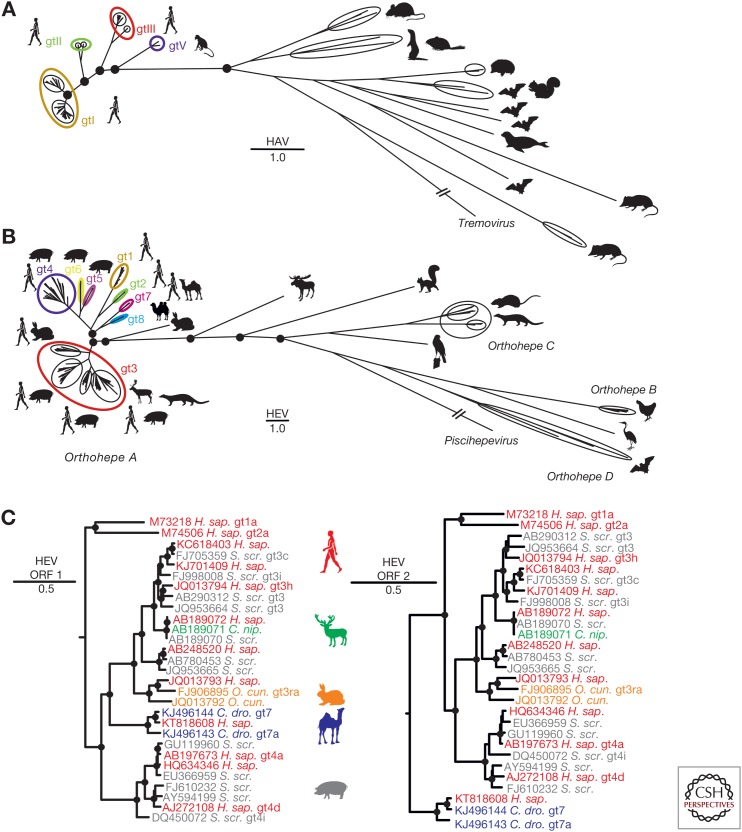
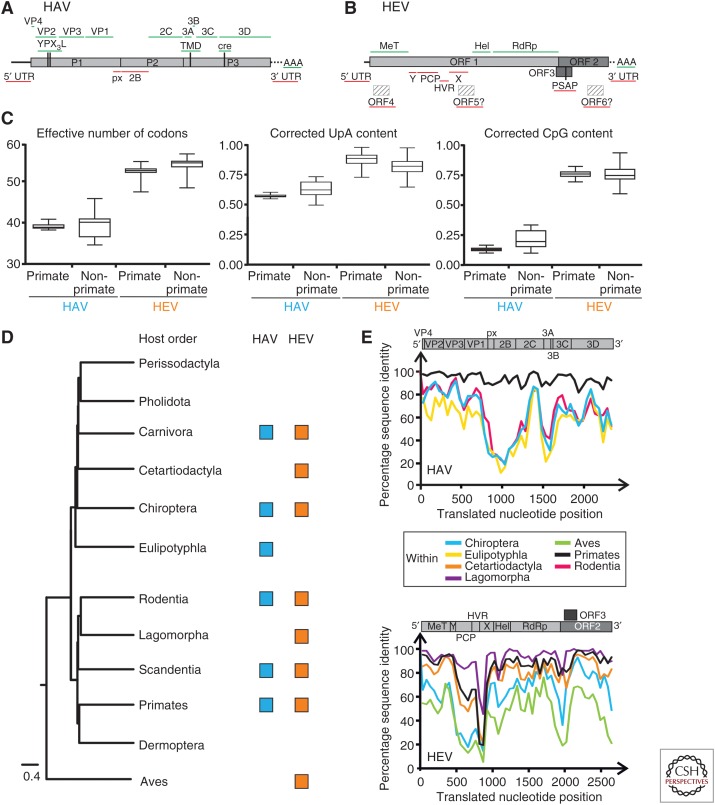
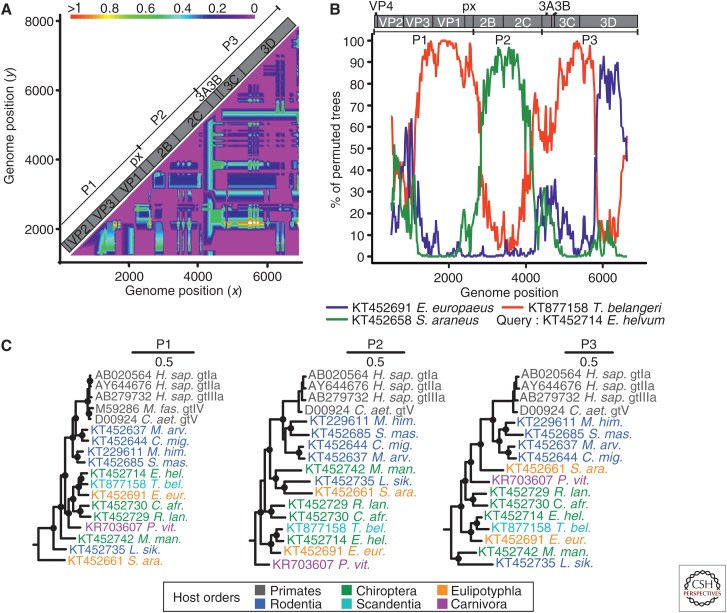
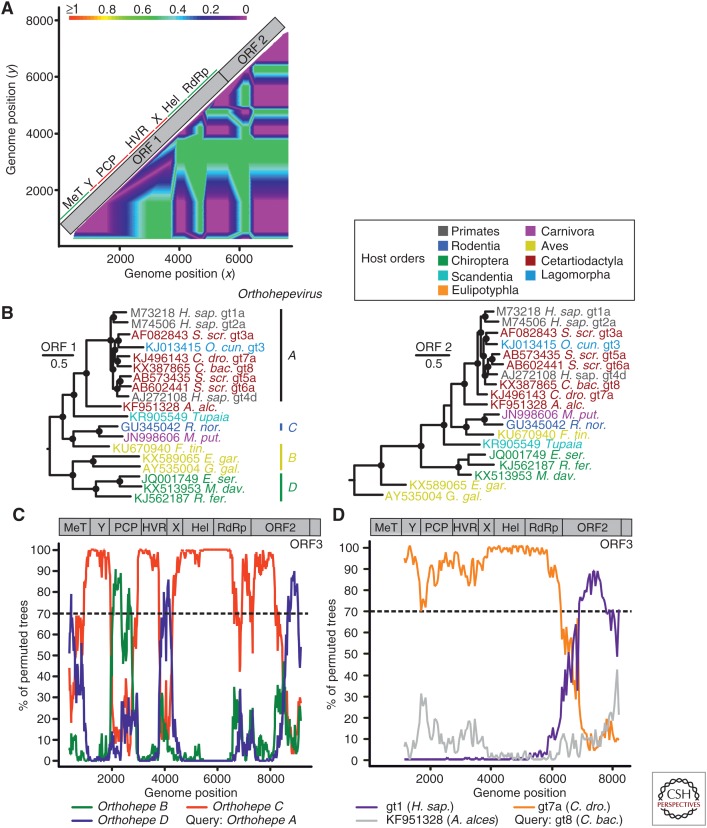
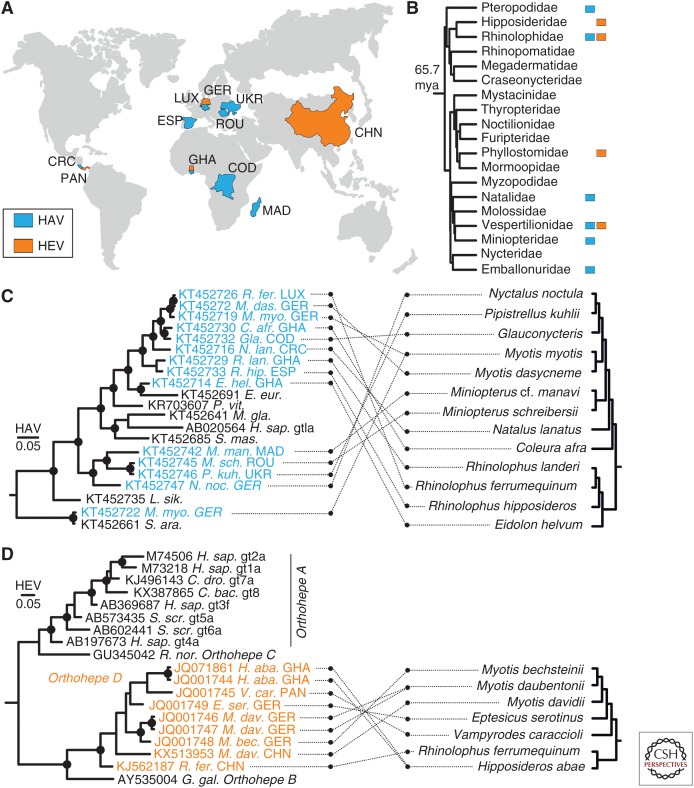
The enterically transmitted hepatitis A (HAV) and hepatitis E viruses (HEV) are the leading causes of acute viral hepatitis in humans. Despite the discovery of HAV and HEV 40-50 years ago, their evolutionary origins remain unclear. Recent discoveries of numerous nonprimate hepatoviruses and hepeviruses allow revisiting the evolutionary history of these viruses. In this review, we provide detailed phylogenomic analyses of primate and nonprimate hepatoviruses and hepeviruses. We identify conserved and divergent genomic properties and corroborate historical interspecies transmissions by phylogenetic comparisons and recombination analyses. We discuss the likely non-recent origins of human HAV and HEV precursors carried by mammals other than primates, and detail current zoonotic HEV infections. The novel nonprimate hepatoviruses and hepeviruses offer exciting new possibilities for future research focusing on host range and the unique biological properties of HAV and HEV.
Copyright © 2018 Cold Spring Harbor Laboratory Press; all rights reserved.
Figures
References
-
- Balayan MS, Andzhaparidze AG, Savinskaya SS, Ketiladze ES, Braginsky DM, Savinov AP, Poleschuk VF. 1983. Evidence for a virus in non-A, non-B hepatitis transmitted via the fecal-oral route. Intervirology 20: 23–31. - PubMed
-
- Batts W, Yun S, Hedrick R, Winton J. 2011. A novel member of the family Hepeviridae from cutthroat trout (Oncorhynchus clarkii). Virus Res 158: 116–123. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical