

Accurate and sensitive quantification of protein-DNA binding affinity
- PMID: 29610332
- PMCID: PMC5910815
- DOI: 10.1073/pnas.1714376115
Accurate and sensitive quantification of protein-DNA binding affinity
Abstract
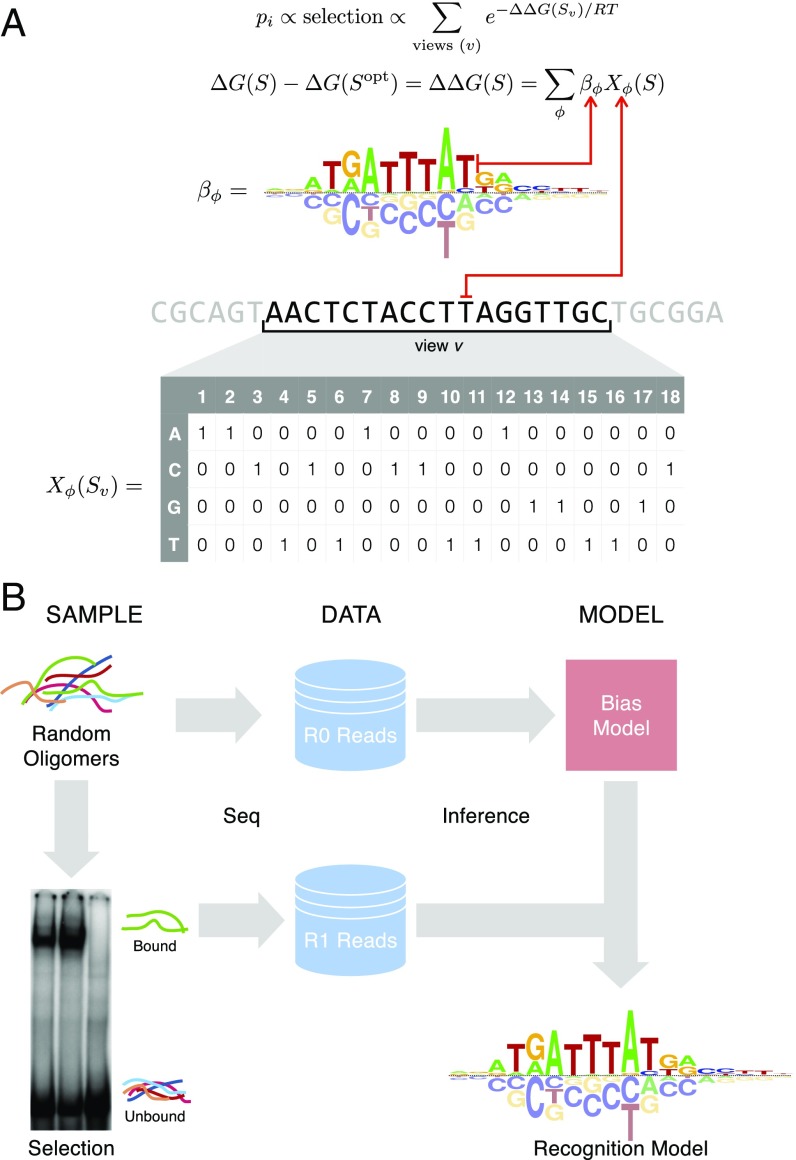
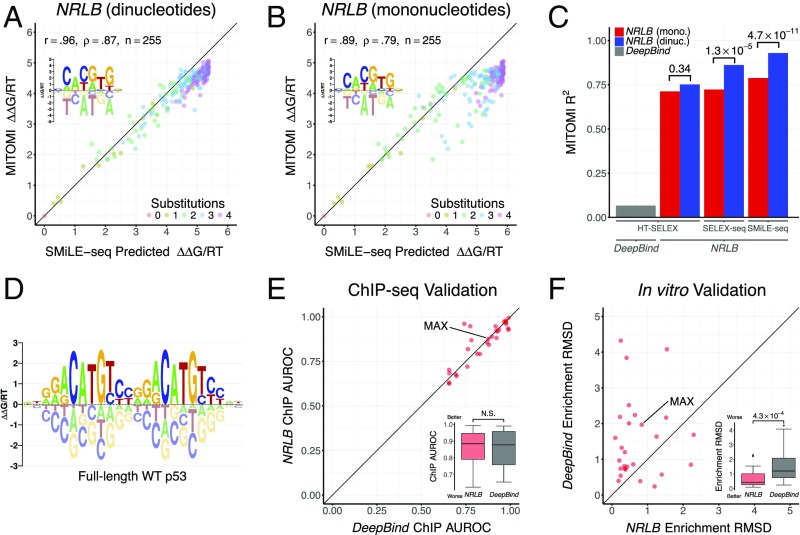
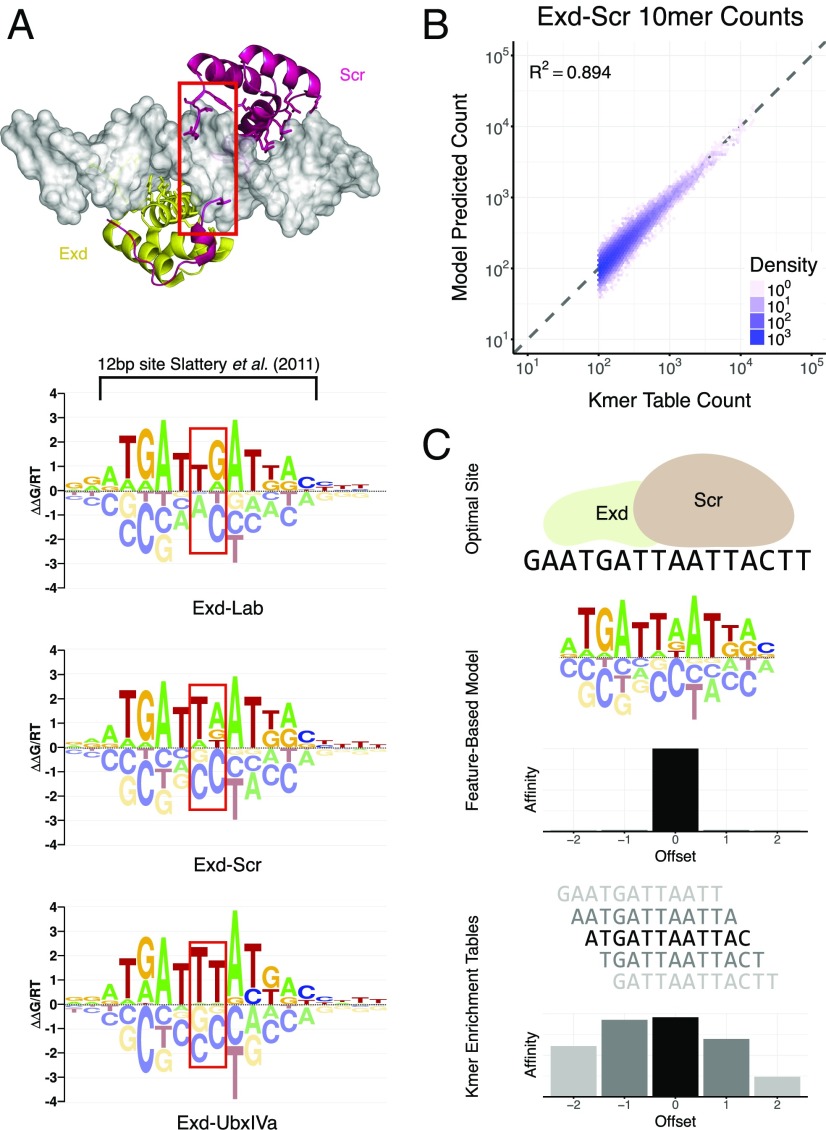
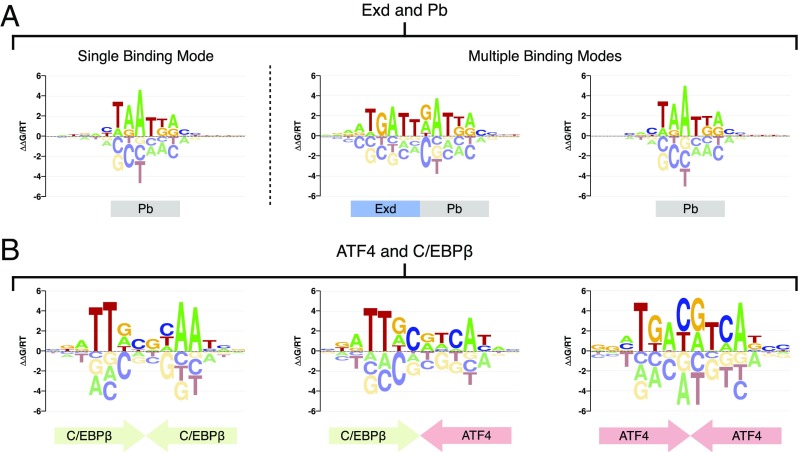
Transcription factors (TFs) control gene expression by binding to genomic DNA in a sequence-specific manner. Mutations in TF binding sites are increasingly found to be associated with human disease, yet we currently lack robust methods to predict these sites. Here, we developed a versatile maximum likelihood framework named No Read Left Behind (NRLB) that infers a biophysical model of protein-DNA recognition across the full affinity range from a library of in vitro selected DNA binding sites. NRLB predicts human Max homodimer binding in near-perfect agreement with existing low-throughput measurements. It can capture the specificity of the p53 tetramer and distinguish multiple binding modes within a single sample. Additionally, we confirm that newly identified low-affinity enhancer binding sites are functional in vivo, and that their contribution to gene expression matches their predicted affinity. Our results establish a powerful paradigm for identifying protein binding sites and interpreting gene regulatory sequences in eukaryotic genomes.
Keywords: SELEX; computational modeling; enhancer assays; low-affinity binding sites; transcription factors.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
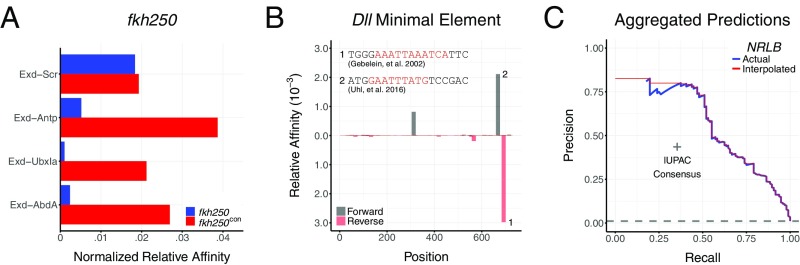
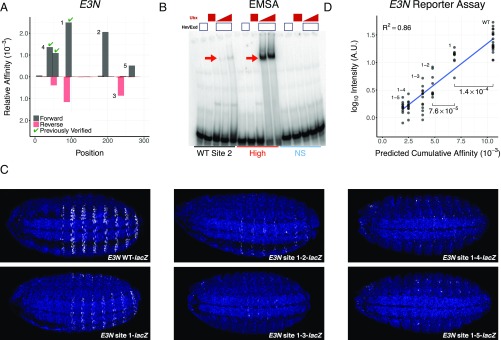
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
