

A global transcriptional network connecting noncoding mutations to changes in tumor gene expression
- PMID: 29610481
- PMCID: PMC5893414
- DOI: 10.1038/s41588-018-0091-2
A global transcriptional network connecting noncoding mutations to changes in tumor gene expression
Abstract
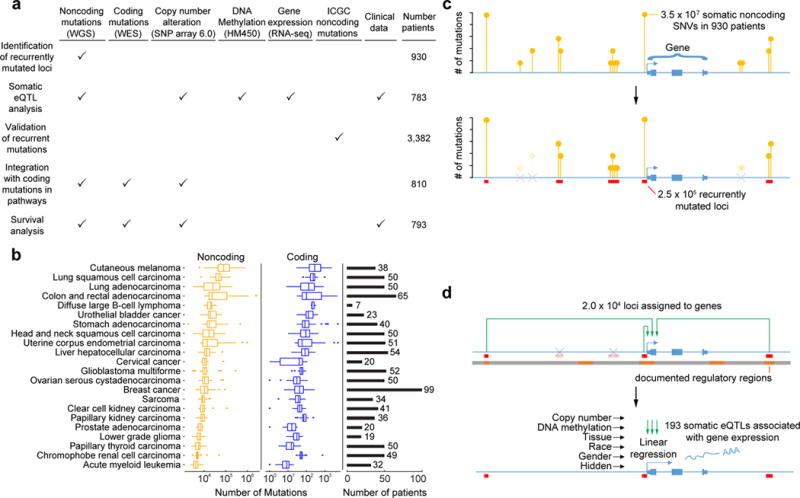
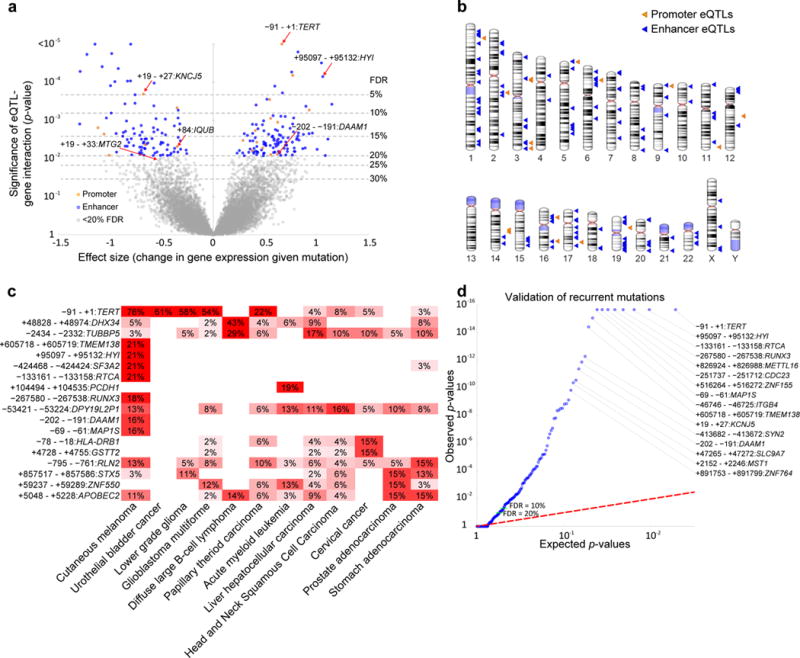
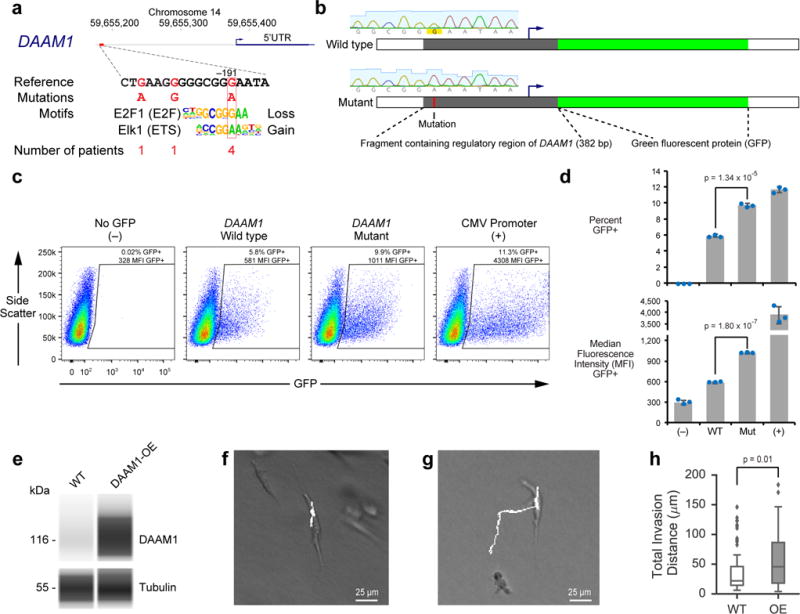
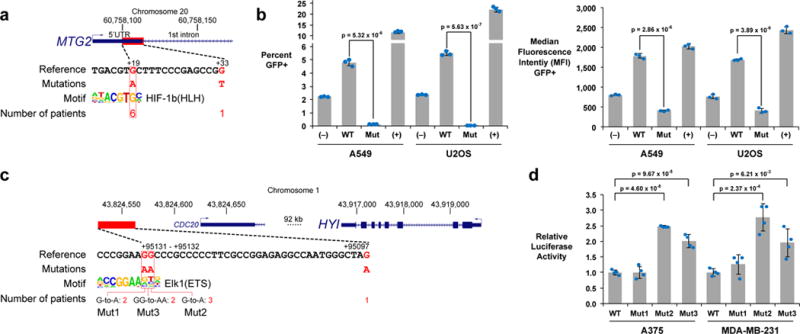
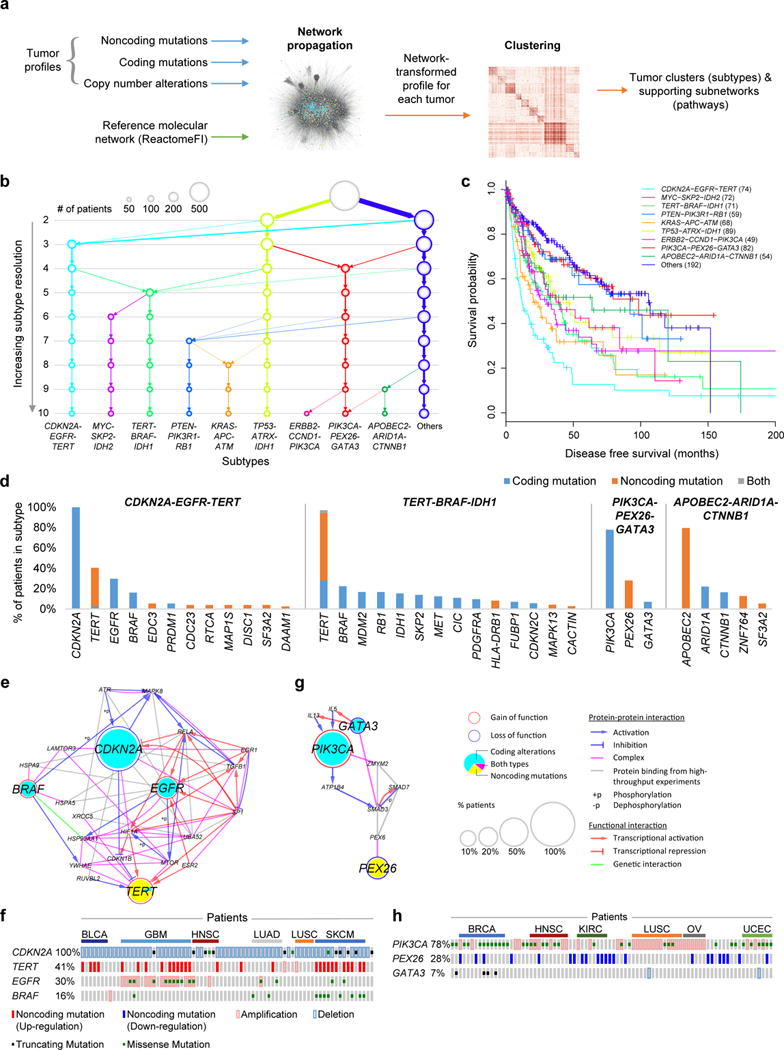
Although cancer genomes are replete with noncoding mutations, the effects of these mutations remain poorly characterized. Here we perform an integrative analysis of 930 tumor whole genomes and matched transcriptomes, identifying a network of 193 noncoding loci in which mutations disrupt target gene expression. These 'somatic eQTLs' (expression quantitative trait loci) are frequently mutated in specific cancer tissues, and the majority can be validated in an independent cohort of 3,382 tumors. Among these, we find that the effects of noncoding mutations on DAAM1, MTG2 and HYI transcription are recapitulated in multiple cancer cell lines and that increasing DAAM1 expression leads to invasive cell migration. Collectively, the noncoding loci converge on a set of core pathways, permitting a classification of tumors into pathway-based subtypes. The somatic eQTL network is disrupted in 88% of tumors, suggesting widespread impact of noncoding mutations in cancer.
Conflict of interest statement
Trey Ideker is co-founder of Data4Cure, Inc. and has an equity interest. Trey Ideker has an equity interest in Ideaya BioSciences, Inc. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict of interest policies. No potential conflicts of interest were disclosed by the other authors.
Figures
References
-
- Khurana E, et al. Role of non-coding sequence variants in cancer. Nat Rev Genet. 2016;17:93–108. - PubMed
Methods-only References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
