

KARS-related diseases: progressive leukoencephalopathy with brainstem and spinal cord calcifications as new phenotype and a review of literature
- PMID: 29615062
- PMCID: PMC5883414
- DOI: 10.1186/s13023-018-0788-4
KARS-related diseases: progressive leukoencephalopathy with brainstem and spinal cord calcifications as new phenotype and a review of literature
Abstract
Background: KARS encodes lysyl- transfer ribonucleic acid (tRNA) synthetase, which catalyzes the aminoacylation of tRNA-Lys in the cytoplasm and mitochondria. Eleven families/sporadic patients and 16 different mutations in KARS have been reported to date. The associated clinical phenotype is heterogeneous ranging from early onset encephalopathy to isolated peripheral neuropathy or nonsyndromic hearing impairment. Recently additional presentations including leukoencephalopathy as predominant cerebral involvement or cardiomyopathy, isolated or associated with muscular and cerebral involvement, have been reported. A progressive Leukoencephalopathy with brainstem and spinal cord calcifications was previously described in a singleton patient and in two siblings, without the identification of the genetic cause. We reported here about a new severe phenotype associated with biallelic KARS mutations and sharing some common points with the other already reported phenotypes, but with a distinct clinical and neuroimaging picture. Review of KARS mutant patients published to date will be also discussed.
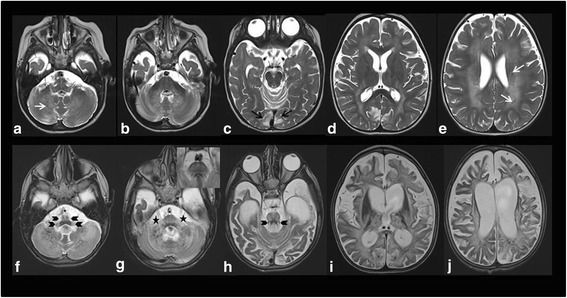
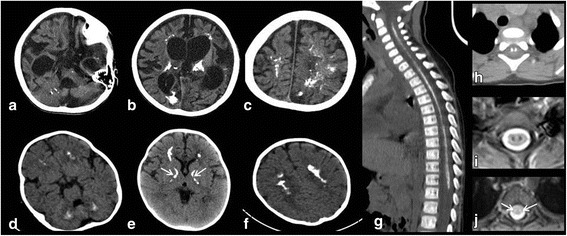
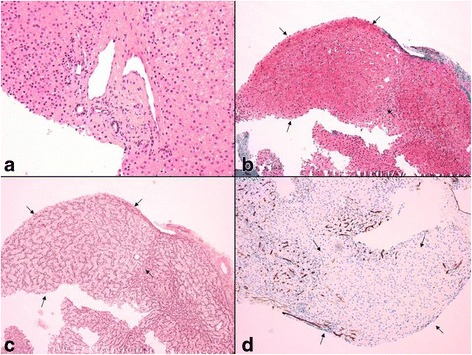
Results: Herein, we report the clinical, biochemical and molecular findings of 2 unreported Italian patients affected by developmental delay, acquired microcephaly, spastic tetraparesis, epilepsy, sensory-neural hypoacusia, visual impairment, microcytic hypochromic anaemia and signs of hepatic dysfunction. MRI pattern in our patients was characterized by progressive diffuse leukoencephalopathy and calcifications extending in cerebral, brainstem and cerebellar white matter, with spinal cord involvement. Genetic analysis performed on these 2 patients and in one subject previously described with similar MRI pattern revealed the presence of biallelic mutations in KARS in all 3 subjects.
Conclusions: With our report we define the molecular basis of the previously described Leukoencephalopathy with Brainstem and Spinal cord Calcification widening the spectrum of KARS related disorders, particularly in childhood onset disease suggestive for mitochondrial impairment. The review of previous cases does not suggest a strict and univocal genotype/phenotype correlation for this highly heterogeneous entity. Moreover, our cases confirm the usefulness of search for common brain and spine MR imaging pattern and of broad genetic screening, in syndromes clinically resembling mitochondrial disorders in spite of normal biochemical assay.
Keywords: Calcifications; KARS; Leukoencephalopathy; Mitochondrial disease.
Conflict of interest statement
Ethics approval and consent to participate
Informed consent for genetic and biochemical studies was obtained from patients’ parents, in agreement with the Declaration of Helsinki and approved by the Ethical Committees of the Neurological Institute “Besta” and Neurological Institute “Mondino”.
Consent for publication
Parents pf pt. A, B and C have given their oral consent to have theclinical data of their sibs published in an anonymized form.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- McMillan HJ, Humphreys P, Smith A, et al. Congenital visual impairment and progressive microcephaly due to Lysyl-transfer ribonucleic acid (RNA) Synthetase (KARS) mutations: the expanding phenotype of aminoacyl-transfer RNA Synthetase mutations in human disease. J Child Neurol. 2015;30(8):1037–1043. doi: 10.1177/0883073814553272. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
