

Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers
- PMID: 29615613
- PMCID: PMC5882811
- DOI: 10.1038/s41467-018-03730-x
Multi-omics analysis reveals neoantigen-independent immune cell infiltration in copy-number driven cancers
Abstract
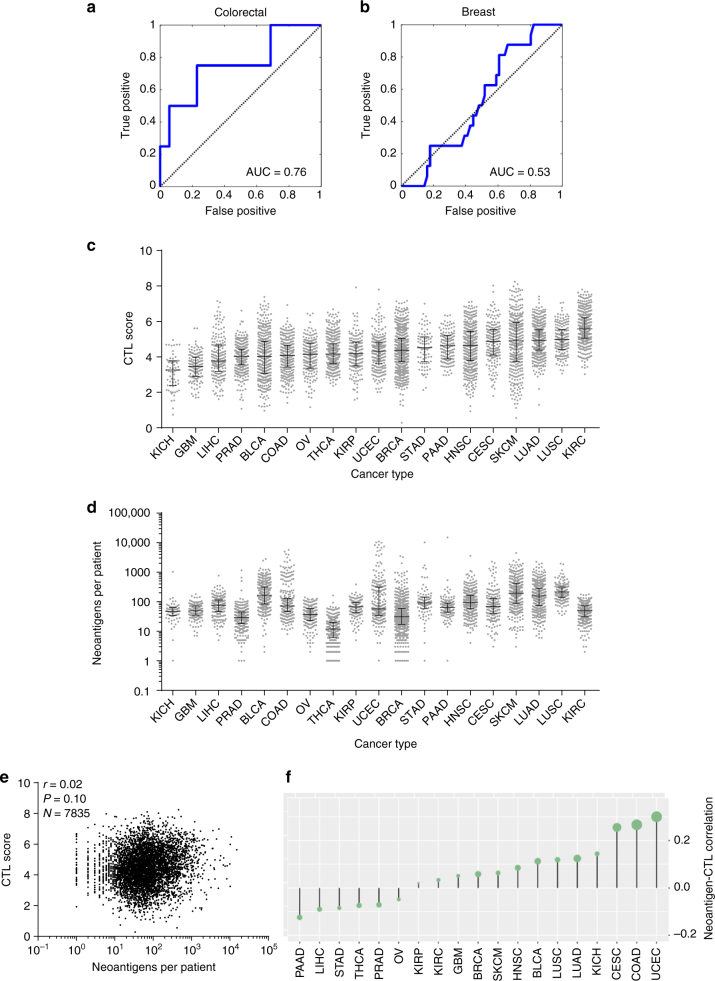
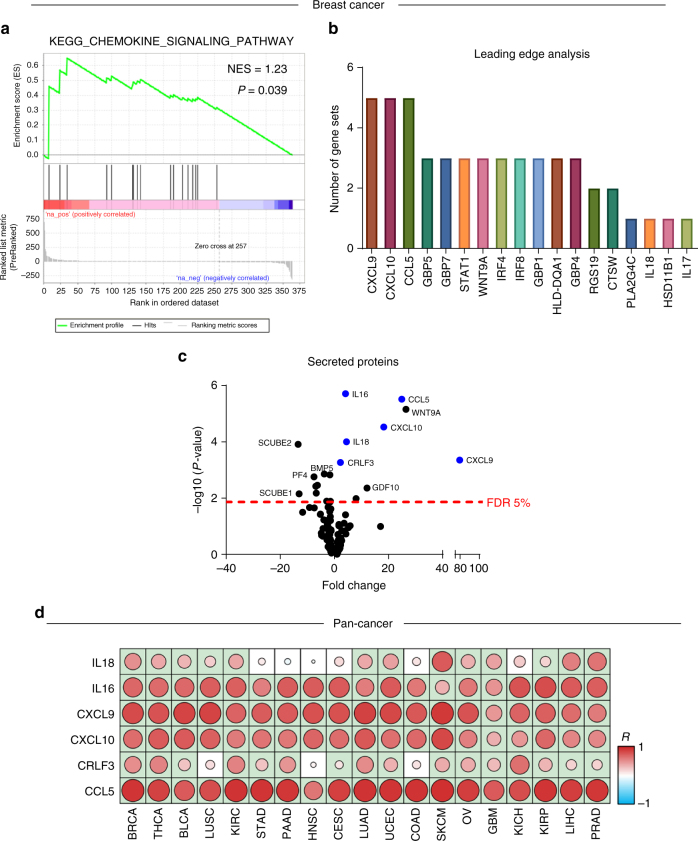
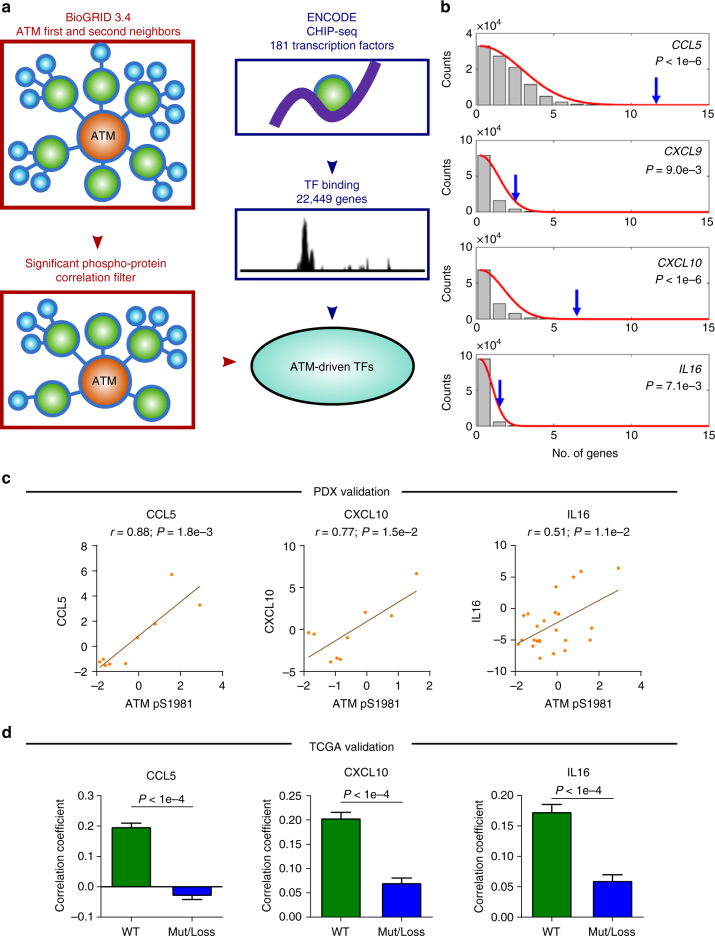
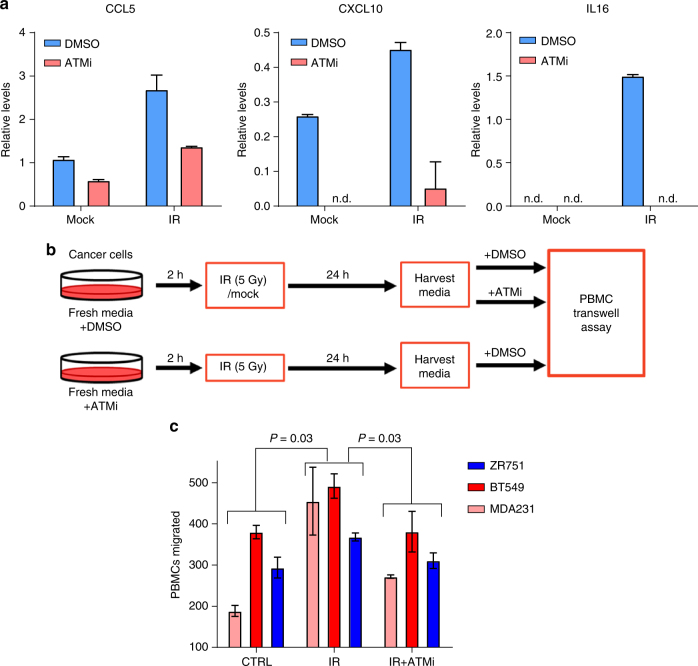
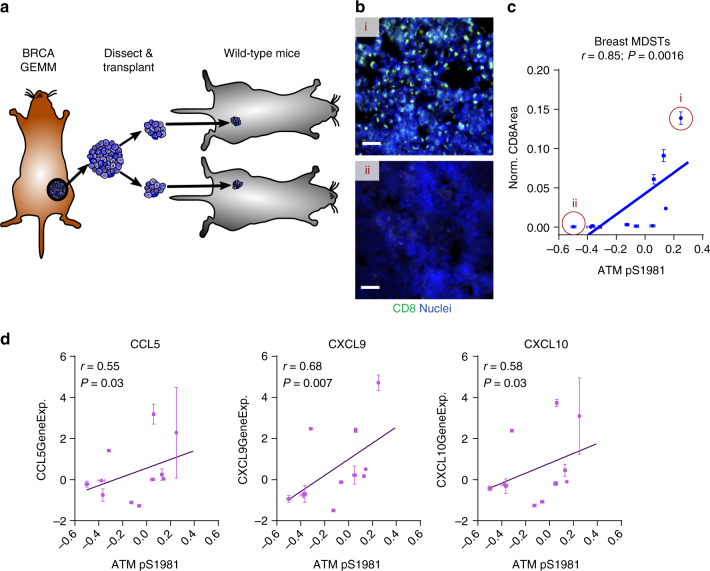
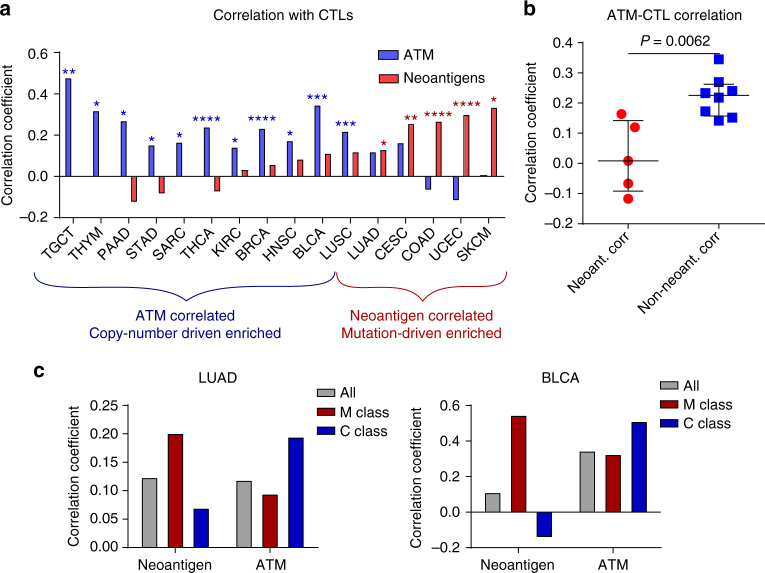
To realize the full potential of immunotherapy, it is critical to understand the drivers of tumor infiltration by immune cells. Previous studies have linked immune infiltration with tumor neoantigen levels, but the broad applicability of this concept remains unknown. Here, we find that while this observation is true across cancers characterized by recurrent mutations, it does not hold for cancers driven by recurrent copy number alterations, such as breast and pancreatic tumors. To understand immune invasion in these cancers, we developed an integrative multi-omics framework, identifying the DNA damage response protein ATM as a driver of cytokine production leading to increased immune infiltration. This prediction was validated in numerous orthogonal datasets, as well as experimentally in vitro and in vivo by cytokine release and immune cell migration. These findings demonstrate diverse drivers of immune cell infiltration across cancer lineages and may facilitate the clinical adaption of immunotherapies across diverse malignancies.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
