

Systematic Analysis of Splice-Site-Creating Mutations in Cancer
- PMID: 29617666
- PMCID: PMC6055527
- DOI: 10.1016/j.celrep.2018.03.052
Systematic Analysis of Splice-Site-Creating Mutations in Cancer
Abstract
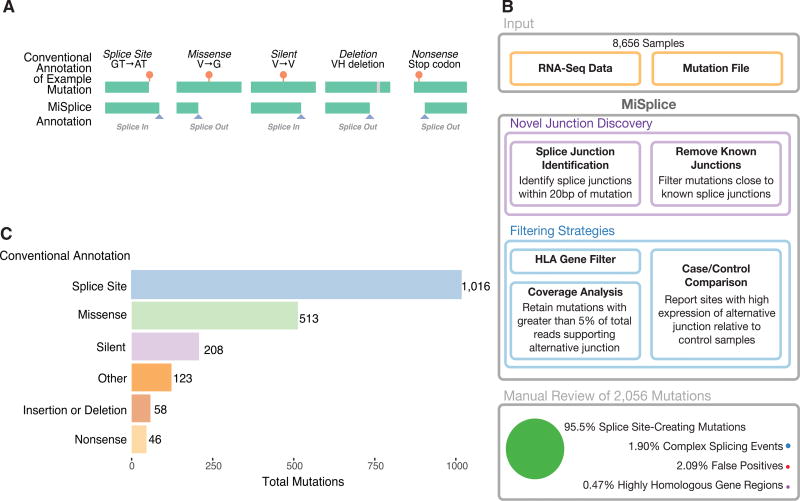
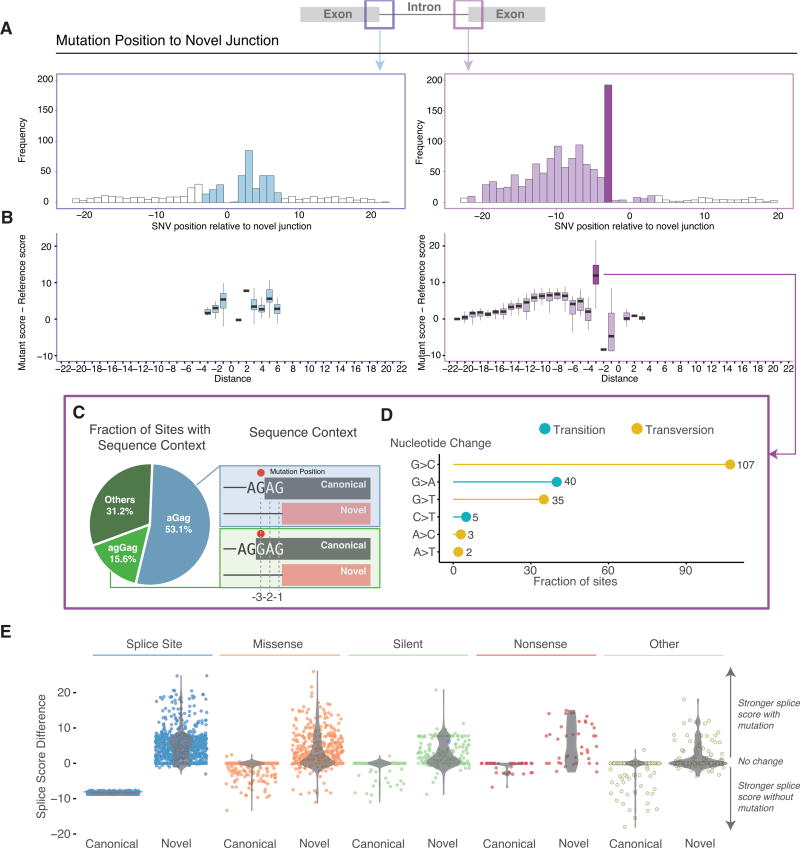
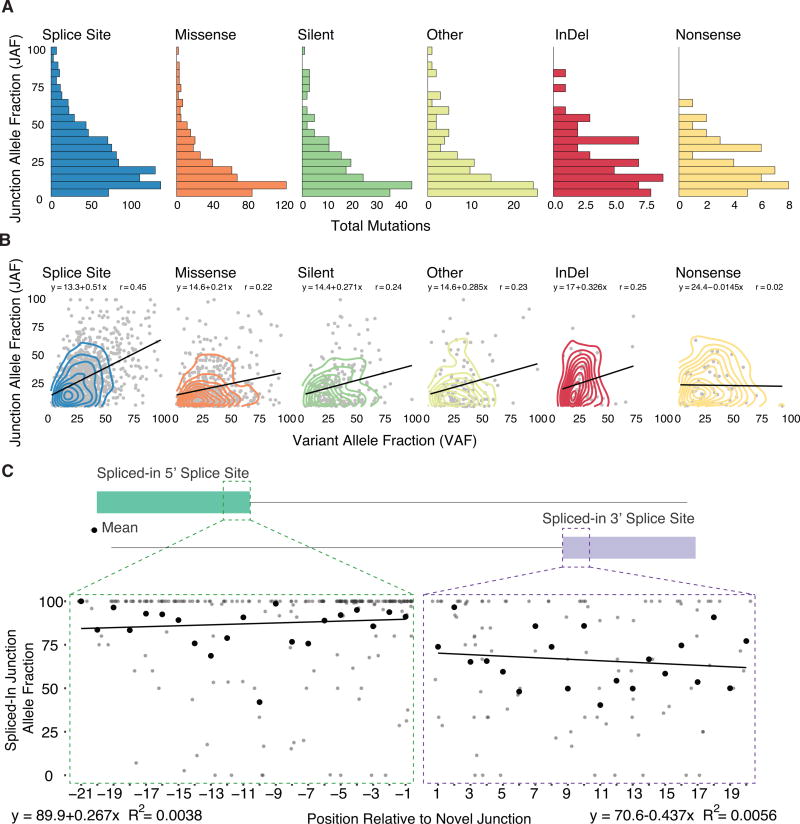
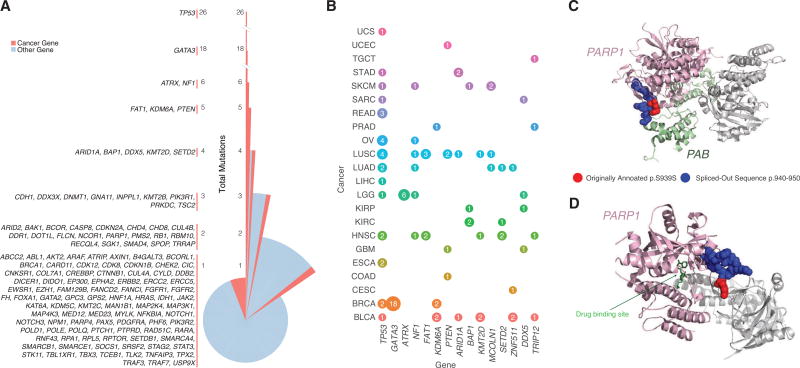
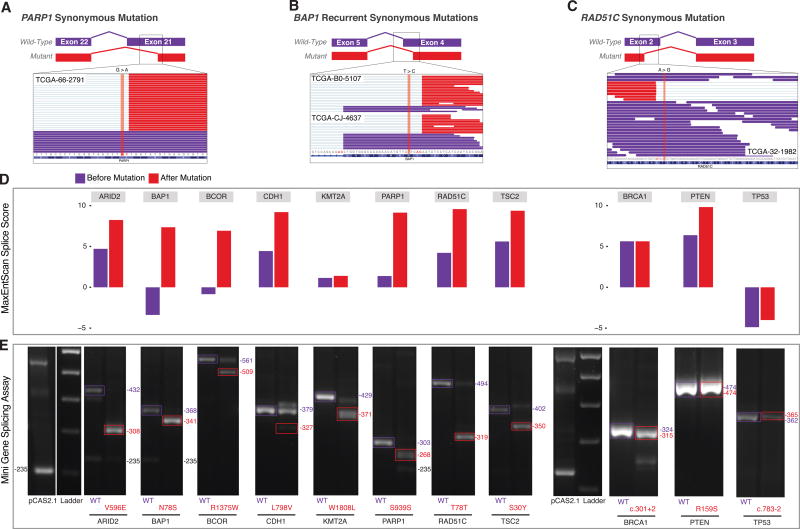
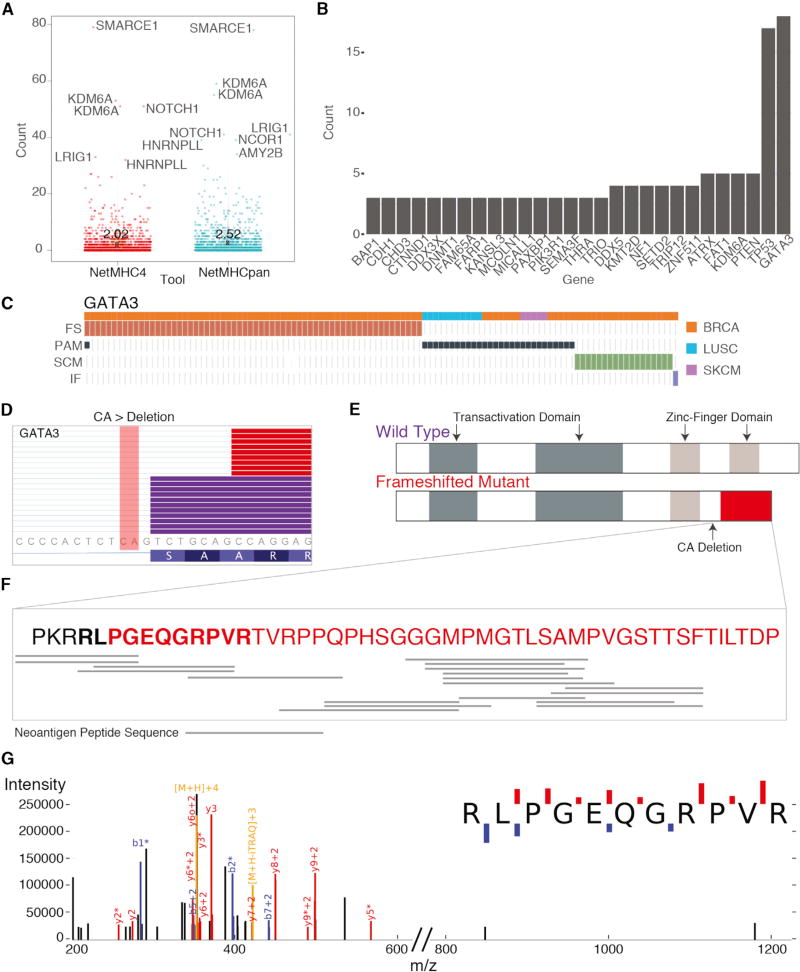
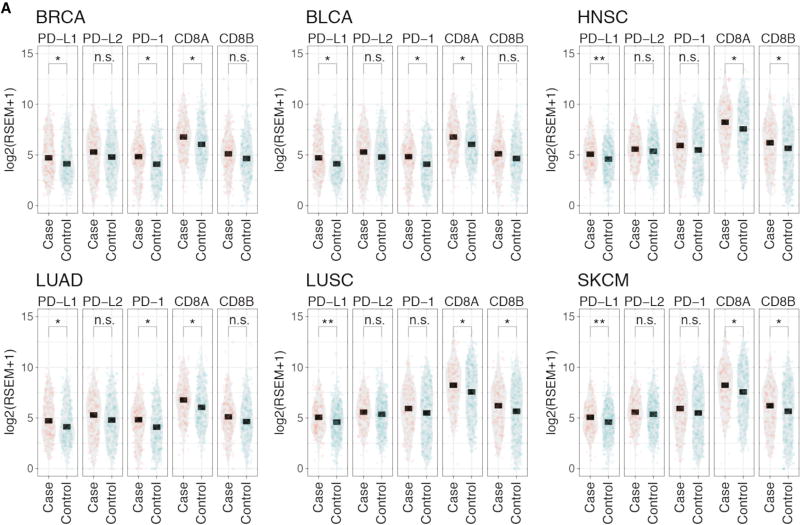
For the past decade, cancer genomic studies have focused on mutations leading to splice-site disruption, overlooking those having splice-creating potential. Here, we applied a bioinformatic tool, MiSplice, for the large-scale discovery of splice-site-creating mutations (SCMs) across 8,656 TCGA tumors. We report 1,964 originally mis-annotated mutations having clear evidence of creating alternative splice junctions. TP53 and GATA3 have 26 and 18 SCMs, respectively, and ATRX has 5 from lower-grade gliomas. Mutations in 11 genes, including PARP1, BRCA1, and BAP1, were experimentally validated for splice-site-creating function. Notably, we found that neoantigens induced by SCMs are likely several folds more immunogenic compared to missense mutations, exemplified by the recurrent GATA3 SCM. Further, high expression of PD-1 and PD-L1 was observed in tumors with SCMs, suggesting candidates for immune blockade therapy. Our work highlights the importance of integrating DNA and RNA data for understanding the functional and the clinical implications of mutations in human diseases.
Keywords: RNA; mutations of clinical relevance; splicing.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Alshammari AH, Shalaby MA, Alanazi MS, Saeed HM. Novel mutations of the PARP-1 gene associated with colorectal cancer in the Saudi population. Asian Pac. J. Cancer Prev. 2014;15:3667–3673. - PubMed
-
- Bonnet C, Krieger S, Vezain M, Rousselin A, Tournier I, Martins A, Berthet P, Chevrier A, Dugast C, Layet V, et al. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J. Med. Genet. 2008;45:438–446. - PubMed
-
- Broeks A, Urbanus JHM, de Knijff P, Devilee P, Nicke M, Klöpper K, Dörk T, Floore AN, van’t Veer LJ. IVS10-6T>G, an ancient ATM germline mutation linked with breast cancer. Hum. Mutat. 2003;21:521–528. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 CA016672/CA/NCI NIH HHS/United States
- U24 CA143882/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- P30 CA016086/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24 CA210972/CA/NCI NIH HHS/United States
- R01 CA180006/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA211006/CA/NCI NIH HHS/United States
- R01 CA163722/CA/NCI NIH HHS/United States
- R01 CA178383/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- R01 HG009711/HG/NHGRI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
