

Optical mapping reveals a higher level of genomic architecture of chained fusions in cancer
- PMID: 29618486
- PMCID: PMC5932612
- DOI: 10.1101/gr.227975.117
Optical mapping reveals a higher level of genomic architecture of chained fusions in cancer
Abstract
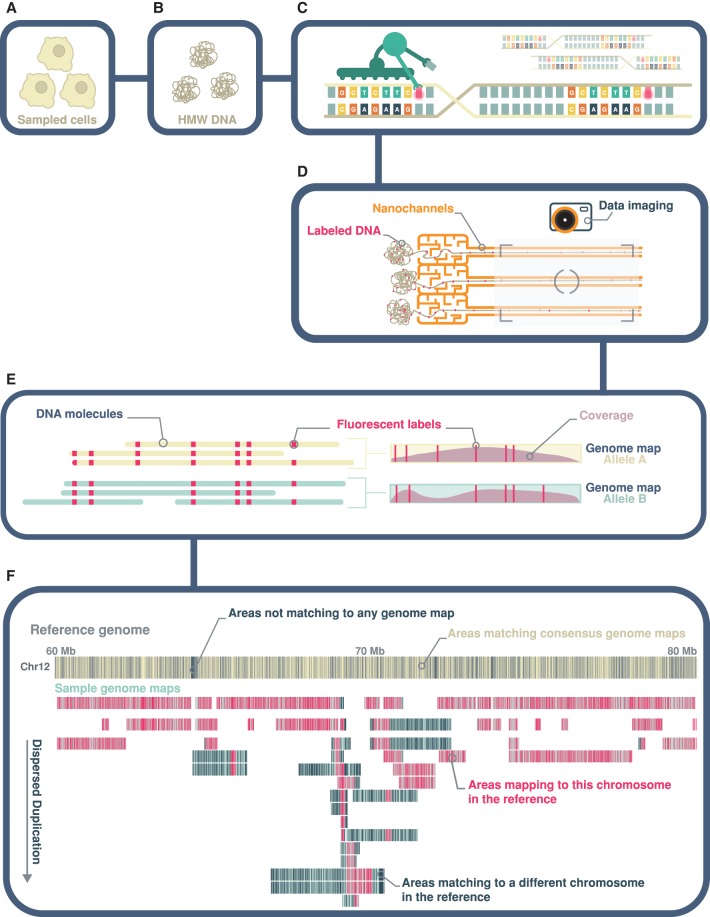
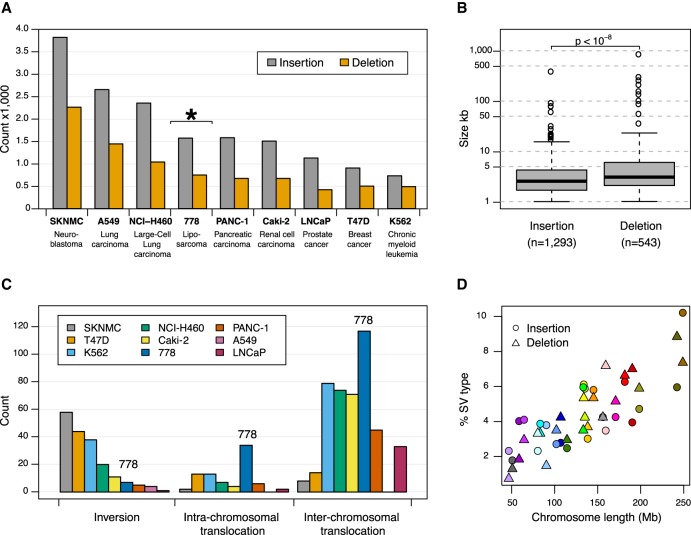
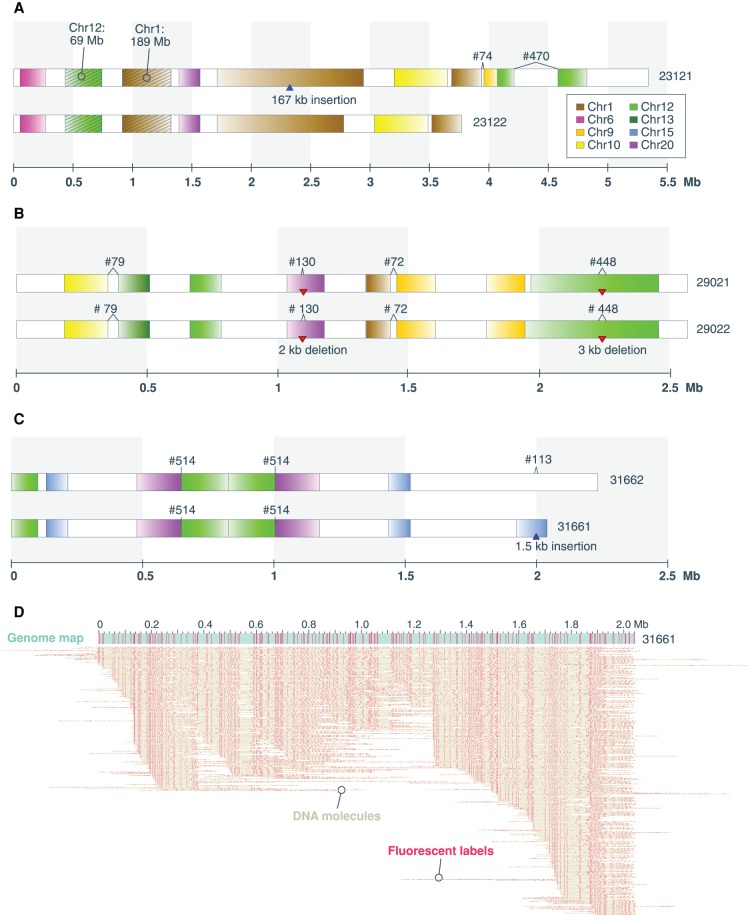
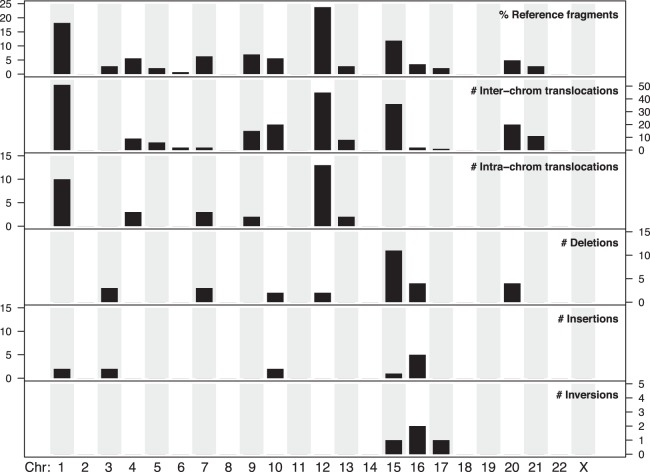
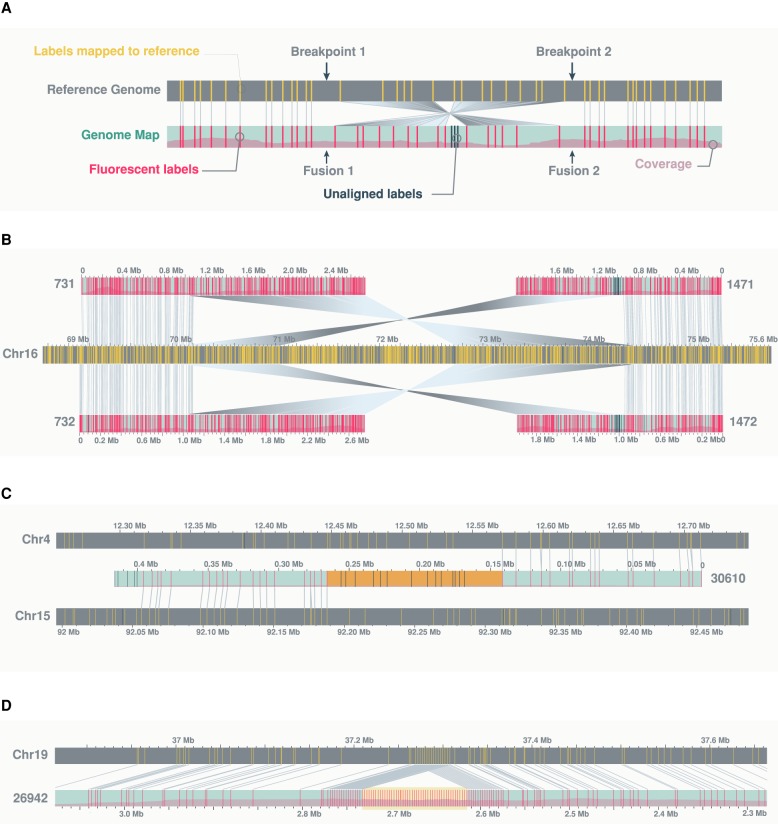
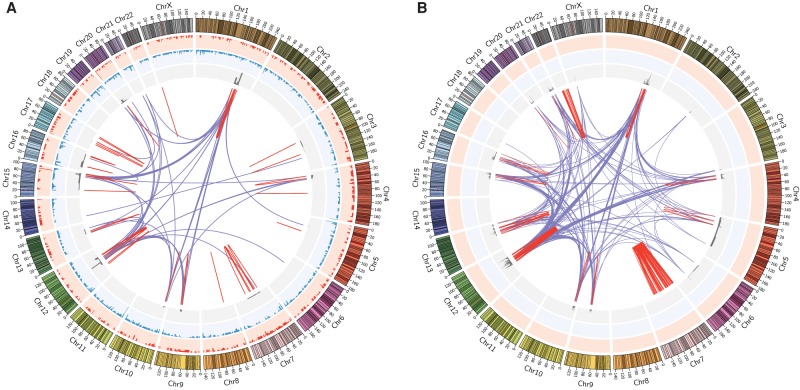
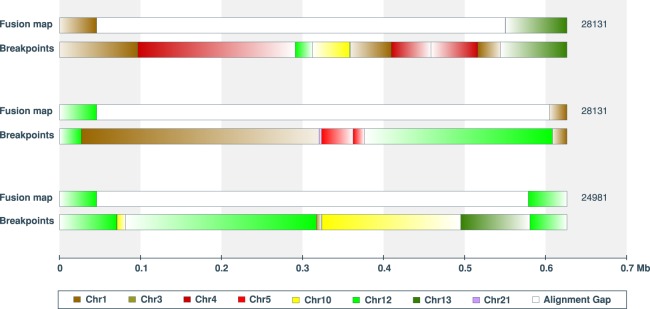
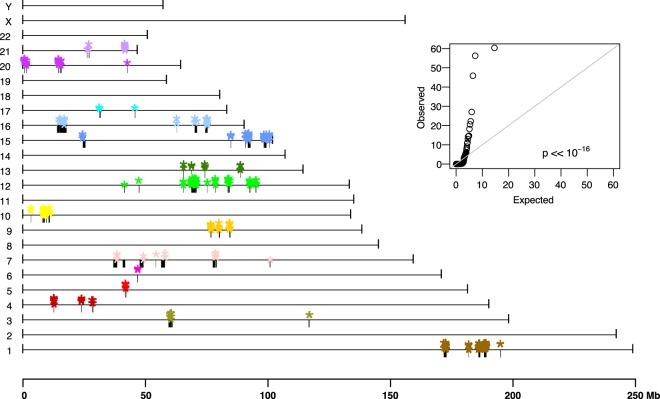
Genomic rearrangements are common in cancer, with demonstrated links to disease progression and treatment response. These rearrangements can be complex, resulting in fusions of multiple chromosomal fragments and generation of derivative chromosomes. Although methods exist for detecting individual fusions, they are generally unable to reconstruct complex chained events. To overcome these limitations, we adopted a new optical mapping approach, allowing megabase-length genome maps to be reconstructed and rearranged genomes to be visualized without loss of integrity. Whole-genome mapping (Bionano Genomics) of a well-studied highly rearranged liposarcoma cell line resulted in 3338 assembled consensus genome maps, including 72 fusion maps. These fusion maps represent 112.3 Mb of highly rearranged genomic regions, illuminating the complex architecture of chained fusions, including content, order, orientation, and size. Spanning the junction of 147 chromosomal translocations, we found a total of 28 Mb of interspersed sequences that could not be aligned to the reference genome. Traversing these interspersed sequences using short-read sequencing breakpoint calls, we were able to identify and place 399 sequencing fragments within the optical mapping gaps, thus illustrating the complementary nature of optical mapping and short-read sequencing. We demonstrate that optical mapping provides a powerful new approach for capturing a higher level of complex genomic architecture, creating a scaffold for renewed interpretation of sequencing data of particular relevance to human cancer.
© 2018 Chan et al.; Published by Cold Spring Harbor Laboratory Press.
Figures

References
-
- Andersson A, Moorman AV, Harrison CJ, Mullighan C. 2016. Acute lymphoblastic leukaemia. In The genetic basis of haematological cancers (ed. Tosi S, Reid AG), pp. 223–264. John Wiley, Chichester, UK.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous