

Defining and Evaluating a Core Genome Multilocus Sequence Typing Scheme for Genome-Wide Typing of Clostridium difficile
- PMID: 29618503
- PMCID: PMC5971537
- DOI: 10.1128/JCM.01987-17
Defining and Evaluating a Core Genome Multilocus Sequence Typing Scheme for Genome-Wide Typing of Clostridium difficile
Abstract
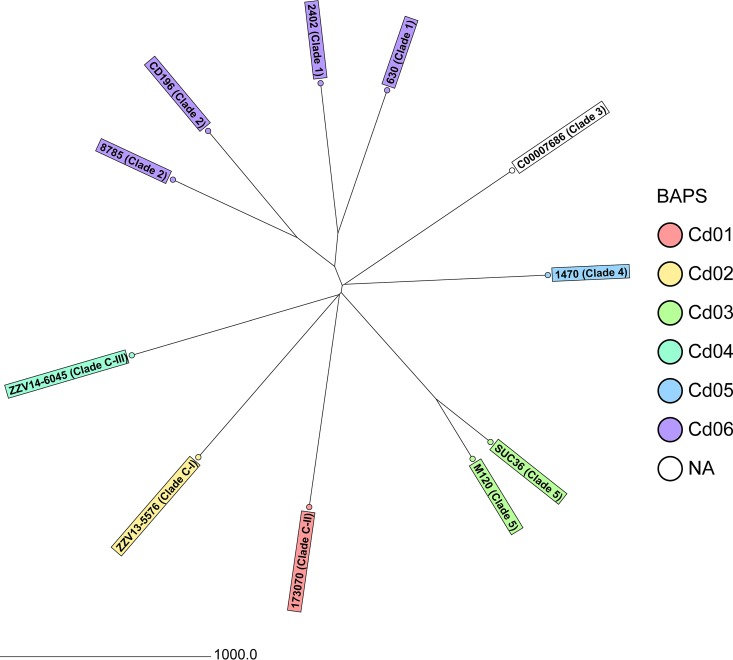
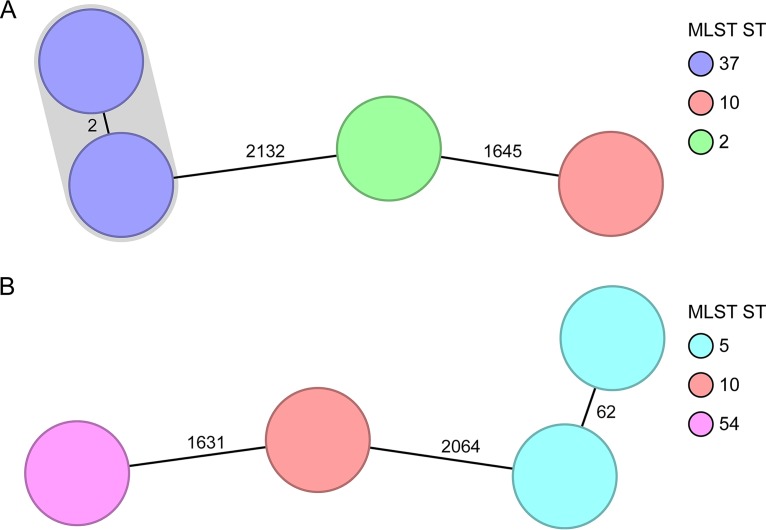
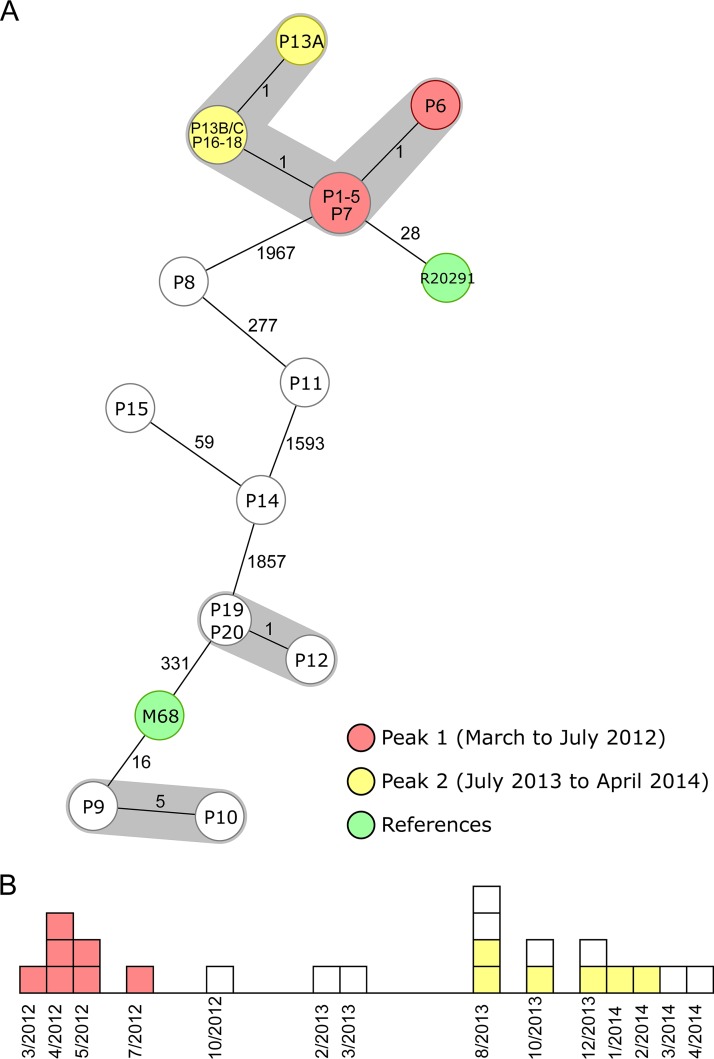
Clostridium difficile, recently renamed Clostridioides difficile, is the most common cause of antibiotic-associated nosocomial gastrointestinal infections worldwide. To differentiate endogenous infections and transmission events, highly discriminatory subtyping is necessary. Today, methods based on whole-genome sequencing data are increasingly used to subtype bacterial pathogens; however, frequently a standardized methodology and typing nomenclature are missing. Here we report a core genome multilocus sequence typing (cgMLST) approach developed for C. difficile Initially, we determined the breadth of the C. difficile population based on all available MLST sequence types with Bayesian inference (BAPS). The resulting BAPS partitions were used in combination with C. difficile clade information to select representative isolates that were subsequently used to define cgMLST target genes. Finally, we evaluated the novel cgMLST scheme with genomes from 3,025 isolates. BAPS grouping (n = 6 groups) together with the clade information led to a total of 11 representative isolates that were included for cgMLST definition and resulted in 2,270 cgMLST genes that were present in all isolates. Overall, 2,184 to 2,268 cgMLST targets were detected in the genome sequences of 70 outbreak-associated and reference strains, and on average 99.3% cgMLST targets (1,116 to 2,270 targets) were present in 2,954 genomes downloaded from the NCBI database, underlining the representativeness of the cgMLST scheme. Moreover, reanalyzing different cluster scenarios with cgMLST were concordant to published single nucleotide variant analyses. In conclusion, the novel cgMLST is representative for the whole C. difficile population, is highly discriminatory in outbreak situations, and provides a unique nomenclature facilitating interlaboratory exchange.
Keywords: Clostridium difficile; cgMLST; typing; whole-genome sequencing.
Copyright © 2018 American Society for Microbiology.
Figures
References
-
- Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:825–834. doi: 10.1056/NEJMoa1408913. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
