

doi: 10.1038/hgv.2018.14.
eCollection 2018.
Novel compound heterozygous variants in the LARP7 gene in a patient with Alazami syndrome
Affiliations
- PMID: 29619239
- PMCID: PMC5874394
- DOI: 10.1038/hgv.2018.14
Item in Clipboard
Novel compound heterozygous variants in the LARP7 gene in a patient with Alazami syndrome
Hum Genome Var.
.
Abstract
The LARP7 gene encodes a chaperone protein of the noncoding RNA 75 K, and mutations in this gene have been identified in patients with Alazami syndrome. Herein, we report another Japanese patient with Alazami syndrome and novel compound heterozygous variants in LARP7 (i.e., c.370delG, p.Glu124fs*38 and c.641_667+25del involving the splice donor site of intron 8). These findings provide further evidence that biallelic LARP7 defects cause the phenotype of Alazami syndrome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
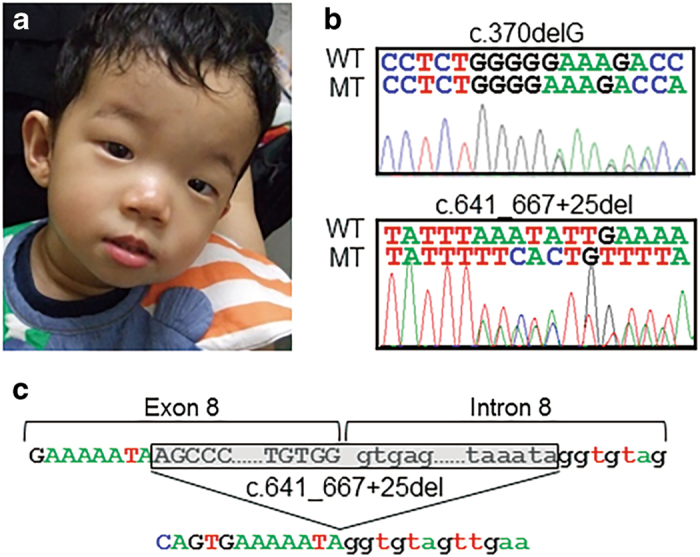
Clinical and genetic findings in the patient. (a) A front view of the patient at 2 years of age showing the distinct facial features of a prominent forehead, narrow palpebral fissures, deep-set eyes, hypertelorism, a broad nose and malar hypoplasia. (b) Electropherograms displaying the two LARP7 mutations in the patient. The upper (c.370delG) and lower (c.641_667+25del) sequences are the sense and antisense sequences, respectively. WT, wild-type allele; MT, mutant allele. (c) A schematic representation of the 52-bp deletion (c.641_667+25del) in LARP7. The exonic and intronic sequences are indicated by capital and lower-case letters, respectively. The deleted sequences are shaded in gray. The deletion involved the splice donor site of intron 8 and was predicted to cause an exon skipping of exon 8.
References
-
- Alazami AM, Al-Owain M, Alzahrani F, Shuaib T, Al-Shamrani H, Al-Falki YH et al. Loss of function mutation in LARP7, chaperone of 7SK ncRNA, causes a syndrome of facial dysmorphism, intellectual disability, and primordial dwarfism. Hum Mutat 2012; 33: 1429–1434. - PubMed
-
- Ling TT, Sorrentino S. Compound heterozygous variants in the LARP7 gene as a cause of Alazami syndrome in a Caucasian female with significant failure to thrive, short stature, and developmental disability. Am J Med Genet A 2016; 170A: 217–219. - PubMed
-
- Hollink IH, Alfadhel M, Al-Wakeel AS, Ababneh F, Pfundt R, de Man SA et al. Broadening the phenotypic spectrum of pathogenic LARP7 variants: two cases with intellectual disability, variable growth retardation and distinct facial features. J Hum Genet 2016; 61: 229–233. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
