

An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome
- PMID: 29619863
- PMCID: PMC5973194
- DOI: 10.1080/19491034.2018.1460045
An overview of treatment strategies for Hutchinson-Gilford Progeria syndrome
Abstract
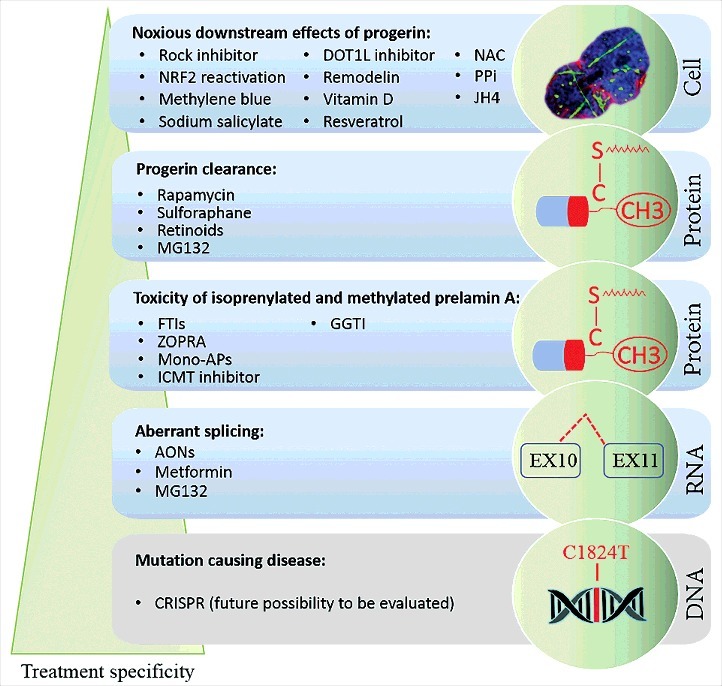
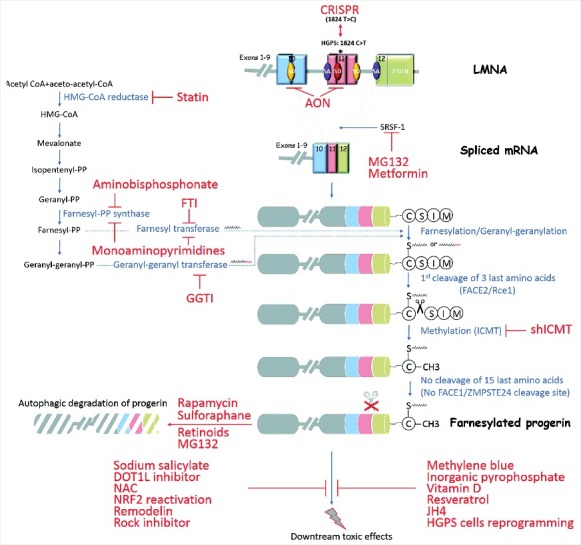
Hutchinson-Gilford progeria syndrome (HGPS) is a sporadic, autosomal dominant disorder characterized by premature and accelerated aging symptoms leading to death at the mean age of 14.6 years usually due to cardiovascular complications. HGPS is caused by a de novo point mutation in the LMNA gene encoding the intermediate filament proteins lamins A and C which are structural components of the nuclear lamina. This mutation leads to the production of a truncated toxic form of lamin A, issued from aberrant splicing and called progerin. Progerin accumulates in HGPS cells' nuclei and is a hallmark of the disease. Small amounts of progerin are also produced during normal aging. HGPS cells and animal preclinical models have provided insights into the molecular and cellular pathways that underlie the disease and have also highlighted possible mechanisms involved in normal aging. This review reports recent medical advances and treatment approaches for patients affected with HGPS.
Keywords: AON; FTI; HGPS; MG132; Metformin; Progerin; Rapamycin; ZOPRA.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous