

Haemochromatosis
- PMID: 29620054
- PMCID: PMC7775623
- DOI: 10.1038/nrdp.2018.16
Haemochromatosis
Abstract
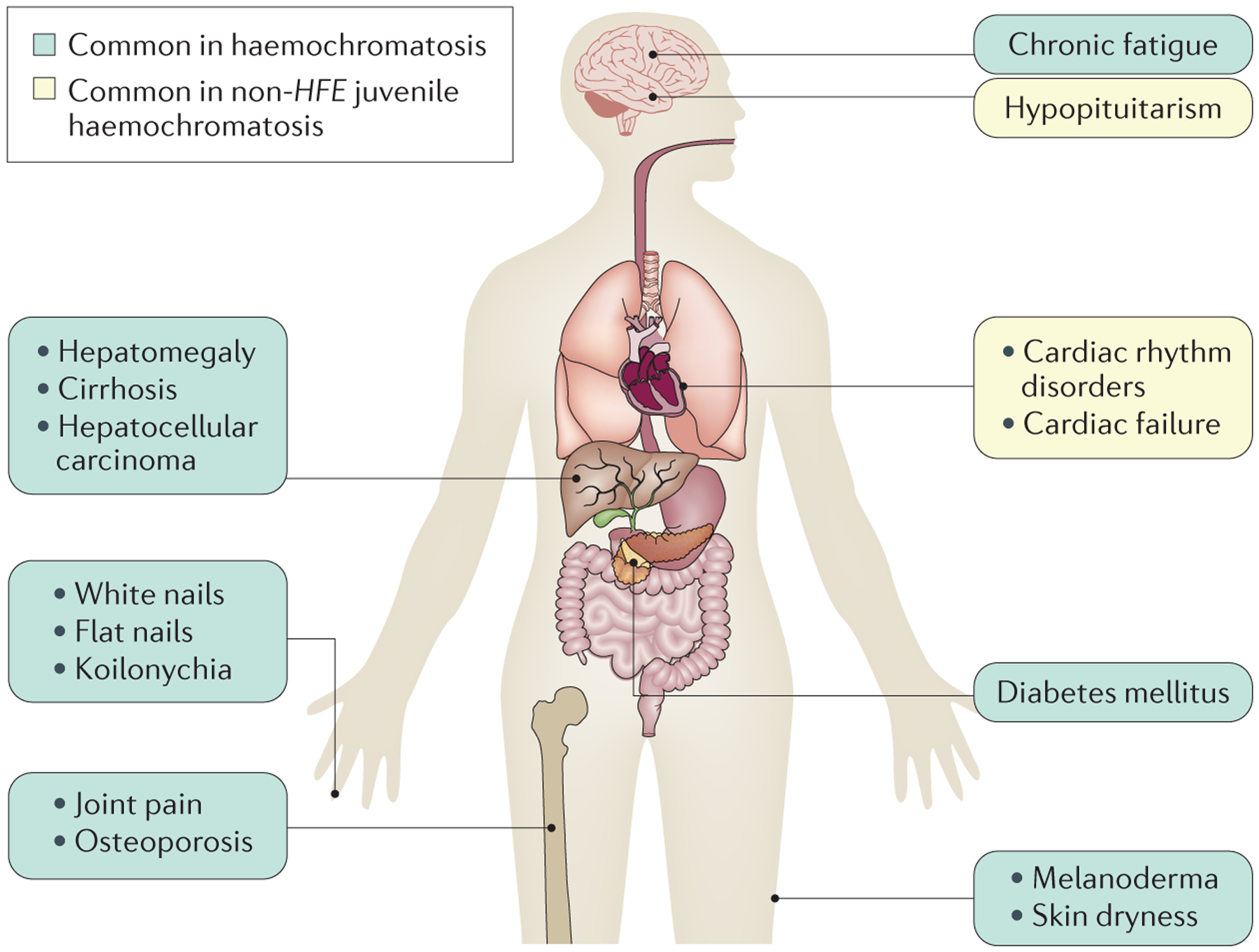
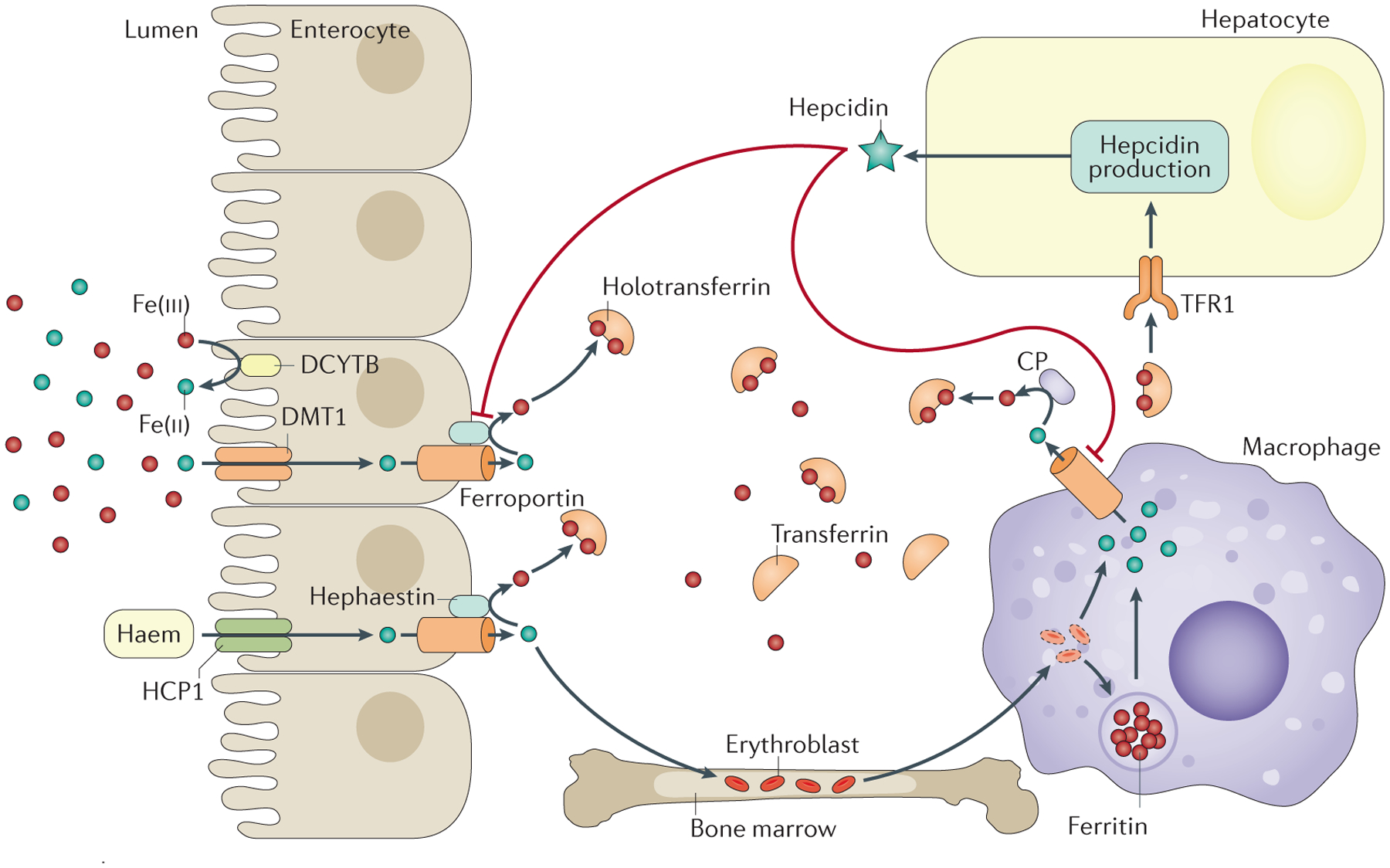
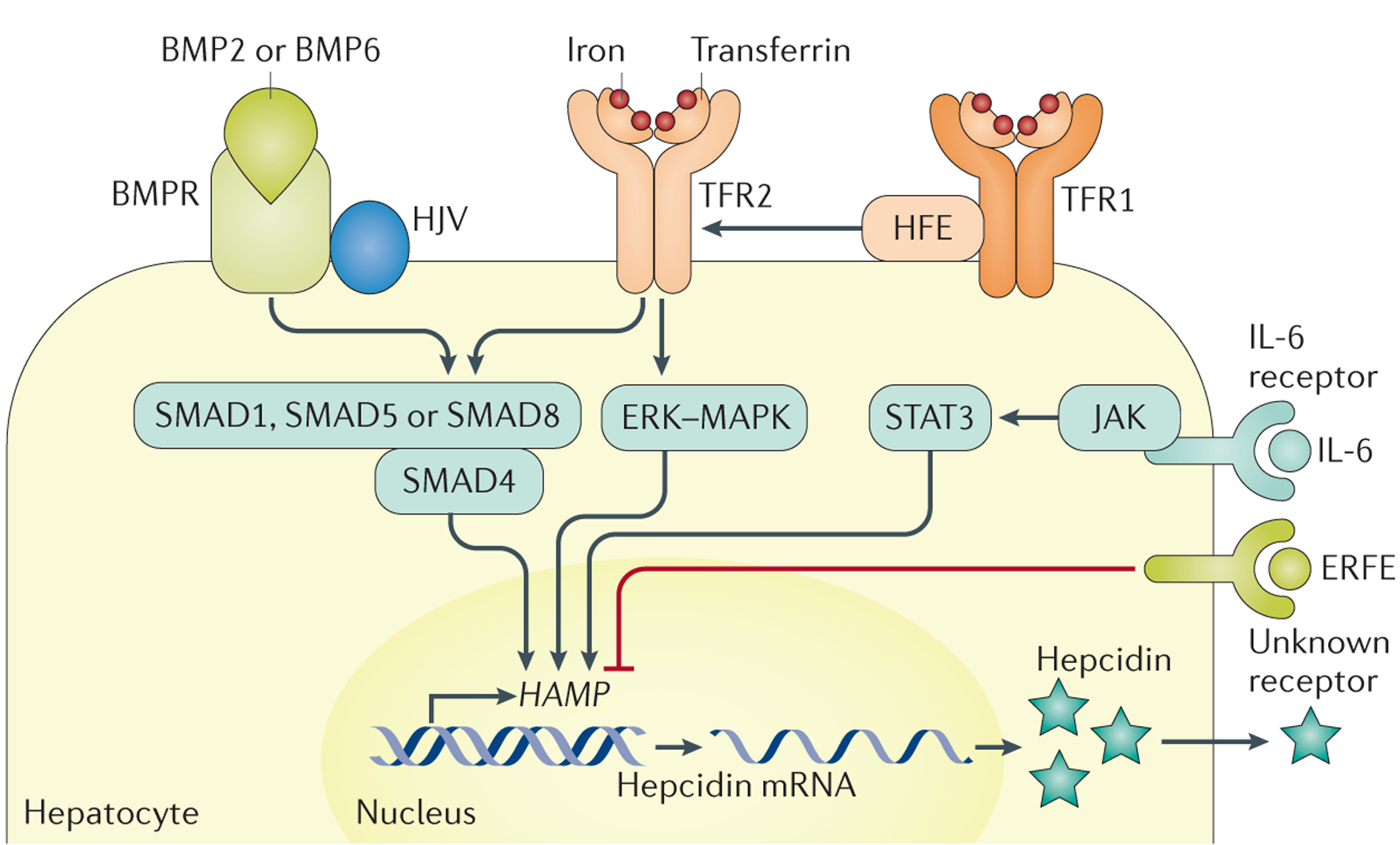
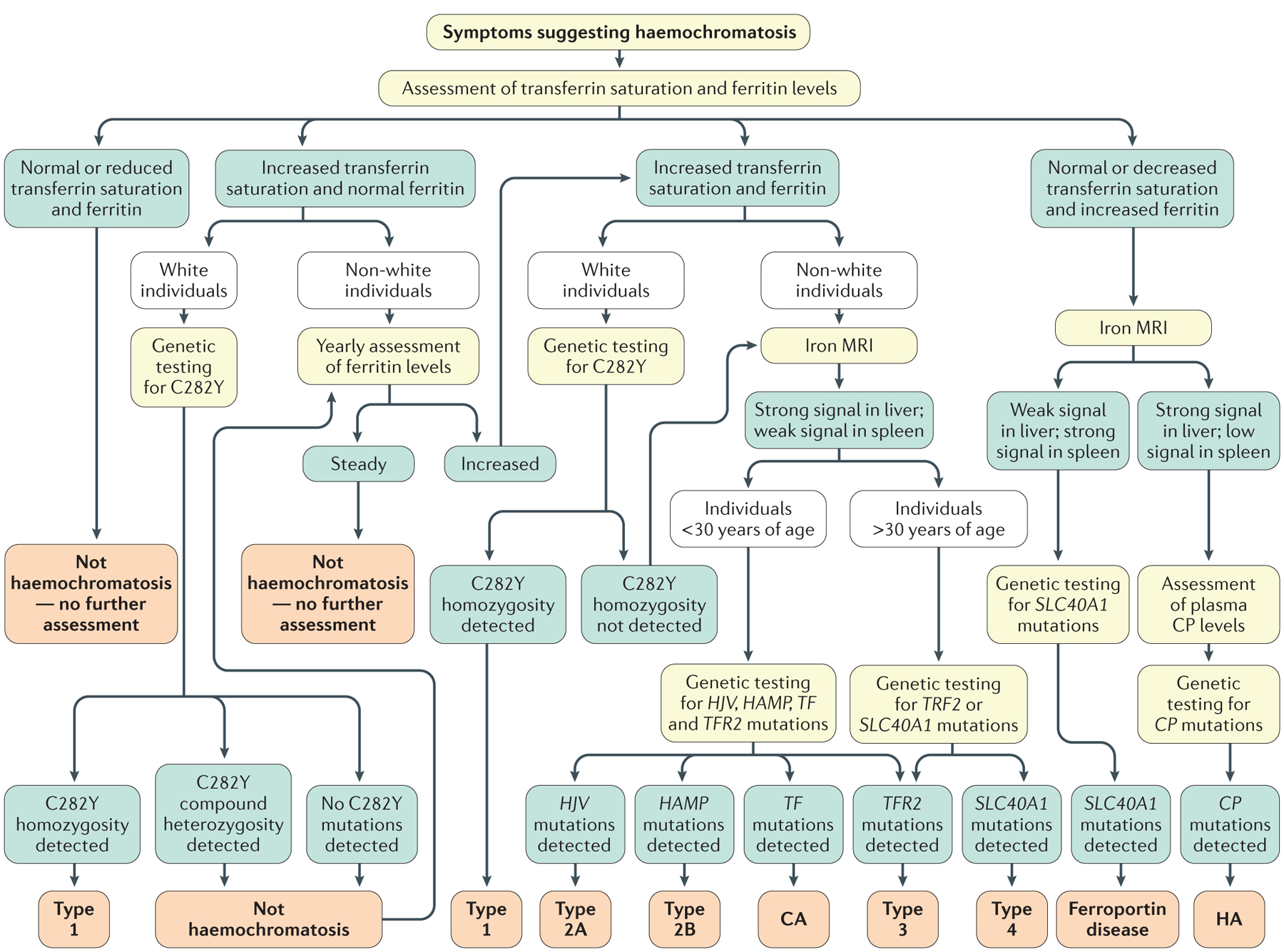
Haemochromatosis is defined as systemic iron overload of genetic origin, caused by a reduction in the concentration of the iron regulatory hormone hepcidin, or a reduction in hepcidin-ferroportin binding. Hepcidin regulates the activity of ferroportin, which is the only identified cellular iron exporter. The most common form of haemochromatosis is due to homozygous mutations (specifically, the C282Y mutation) in HFE, which encodes hereditary haemochromatosis protein. Non-HFE forms of haemochromatosis due to mutations in HAMP, HJV or TFR2 are much rarer. Mutations in SLC40A1 (also known as FPN1; encoding ferroportin) that prevent hepcidin-ferroportin binding also cause haemochromatosis. Cellular iron excess in HFE and non-HFE forms of haemochromatosis is caused by increased concentrations of plasma iron, which can lead to the accumulation of iron in parenchymal cells, particularly hepatocytes, pancreatic cells and cardiomyocytes. Diagnosis is noninvasive and includes clinical examination, assessment of plasma iron parameters, imaging and genetic testing. The mainstay therapy is phlebotomy, although iron chelation can be used in some patients. Hepcidin supplementation might be an innovative future approach.
Conflict of interest statement
Competing interests
P.B. has received lecture fees from Novartis and consulting fees from Novartis and La Jolla Pharmaceutical Company. A.P. has received lecture fees from Novartis, and consulting fees from Novartis, La Jolla Pharmaceutical Company and Mitsubishi Tanabe Pharma Corporation. O.L. has received a research grant from Novartis. All other authors declare no competing interests.
Figures
References
-
- Merryweather-Clarke AT, Pointon JJ, Jouanolle AM, Rochette J & Robson KJ Geography of HFE C282Y and H63D mutations. Genet. Test 4, 183–198 (2000). - PubMed
-
- McLaren CE et al. Hemochromatosis and Iron Overload Screening (HEIRS) study design for an evaluation of 100,000 primary care-based adults. Am. J. Med. Sci 325, 53–62 (2003). - PubMed
-
- Adams PC et al. Hemochromatosis and iron-overload screening in a racially diverse population. N. Engl. J. Med 352, 1769–1778 (2005). - PubMed
-
- Kirk L et al. Haemochromatosis gene frequency in a control and diabetic Irish population. Ir. J. Med. Sci 178, 39–42 (2009). - PubMed
-
- Hanson EH, Imperatore G & Burke W HFE gene and hereditary hemochromatosis: a HuGE review. Hum. Genome Epidemiol. Am. J. Epidemiol 154, 193–206 (2001). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
