

Regulatory role of calpain in neuronal death
- PMID: 29623944
- PMCID: PMC5900522
- DOI: 10.4103/1673-5374.228762
Regulatory role of calpain in neuronal death
Abstract
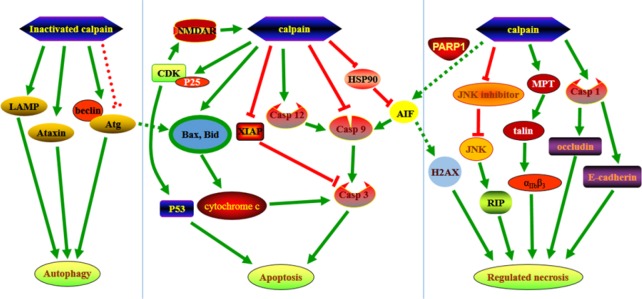
Calpains are a group of calcium-dependent proteases that are over activated by increased intracellular calcium levels under pathological conditions. A wide range of substrates that regulate necrotic, apoptotic and autophagic pathways are affected by calpain. Calpain plays a very important role in neuronal death and various neurological disorders. This review introduces recent research progress related to the regulatory mechanisms of calpain in neuronal death. Various neuronal programmed death pathways including apoptosis, autophagy and regulated necrosis can be divided into receptor interacting protein-dependent necroptosis, mitochondrial permeability transition-dependent necrosis, pyroptosis and poly (ADP-ribose) polymerase 1-mediated parthanatos. Calpains cleave series of key substrates that may lead to cell death or participate in cell death. Regarding the investigation of calpain-mediated programed cell death, it is necessary to identify specific inhibitors that inhibit calpain mediated neuronal death and nervous system diseases.
Keywords: B-cell lymphoma; apoptosis; autophagy; calpain; calpastatin; central nervous system; cyclin-dependent kinases; mitochondrial permeability transition; nerve regeneration; neural regeneration.
Conflict of interest statement
None declared
Figures
References
-
- Alvira D, Ferrer I, Gutierrez-Cuesta J, Garcia-Castro B, Pallas M, Camins A. Activation of the calpain/cdk5/p25 pathway in the girus cinguli in Parkinson’s disease. Parkinsonism Relat D. 2008;14:309–313. - PubMed
-
- Arisan ED, Obakan P, Coker-Gurkan A, Calcabrini A, Agostinelli E, Unsal NP. CDK inhibitors induce mitochondria-mediated apoptosis through the activation of polyamine catabolic pathway in LNCaP, DU145 and PC3 prostate cancer cells. Curr Pharm Design. 2014;20:180–188. - PubMed
-
- Arrington DD, Van Vleet TR, Schnellmann RG. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am J Physiol Cell Physiol. 2006;291:C1159–1171. - PubMed
-
- Bakshi A, Keck CA, Koshkin VS, LeBold DG, Siman R, Snyder EY, McIntosh TK. Caspase-mediated cell death predominates following engraftment of neural progenitor cells into traumatically injured rat brain. Brain Res. 2005;1065:8–19. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
