

A comprehensive and quantitative exploration of thousands of viral genomes
- PMID: 29624169
- PMCID: PMC5908442
- DOI: 10.7554/eLife.31955
A comprehensive and quantitative exploration of thousands of viral genomes
Abstract
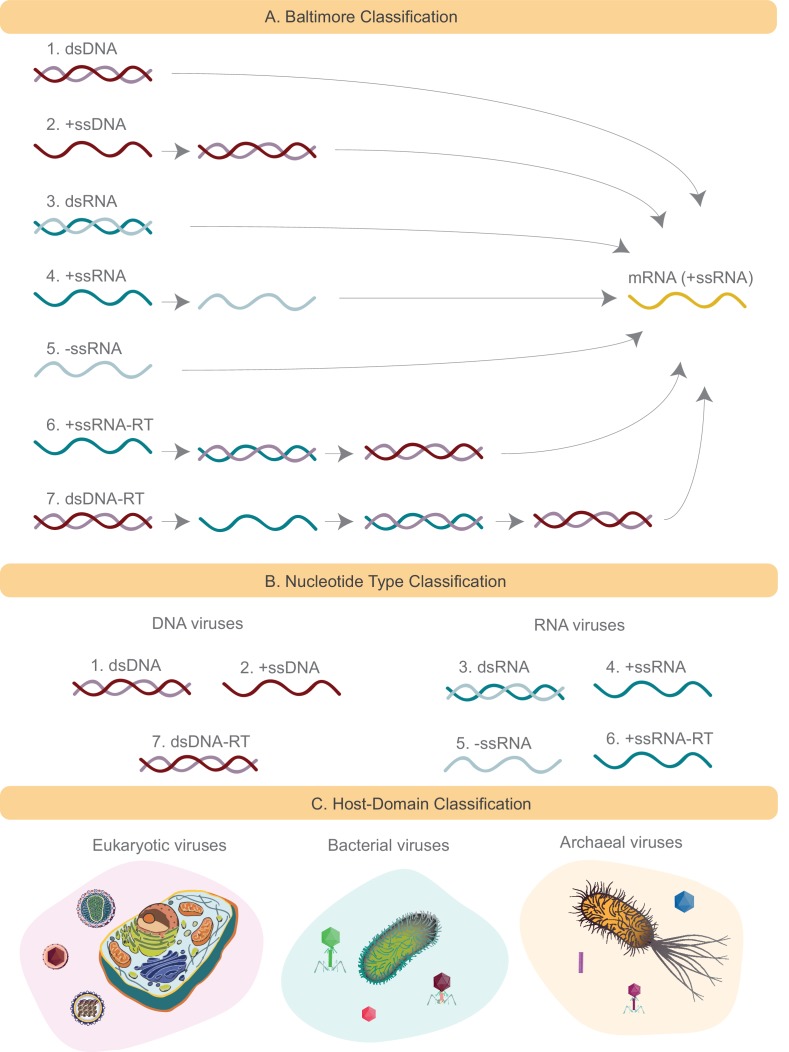
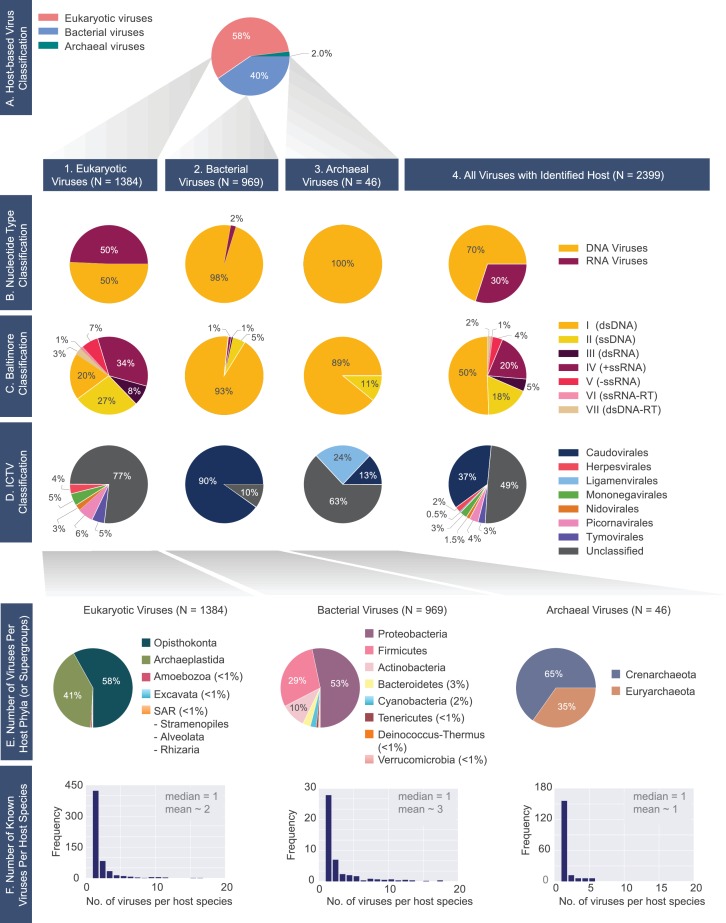
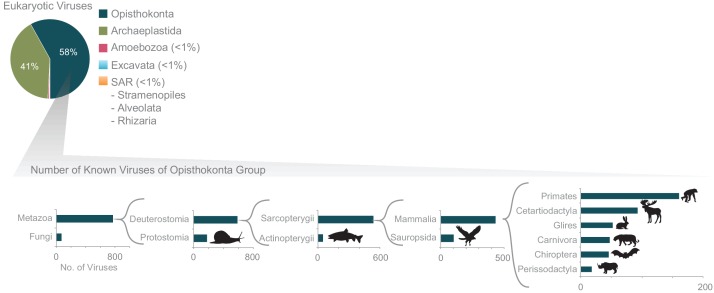
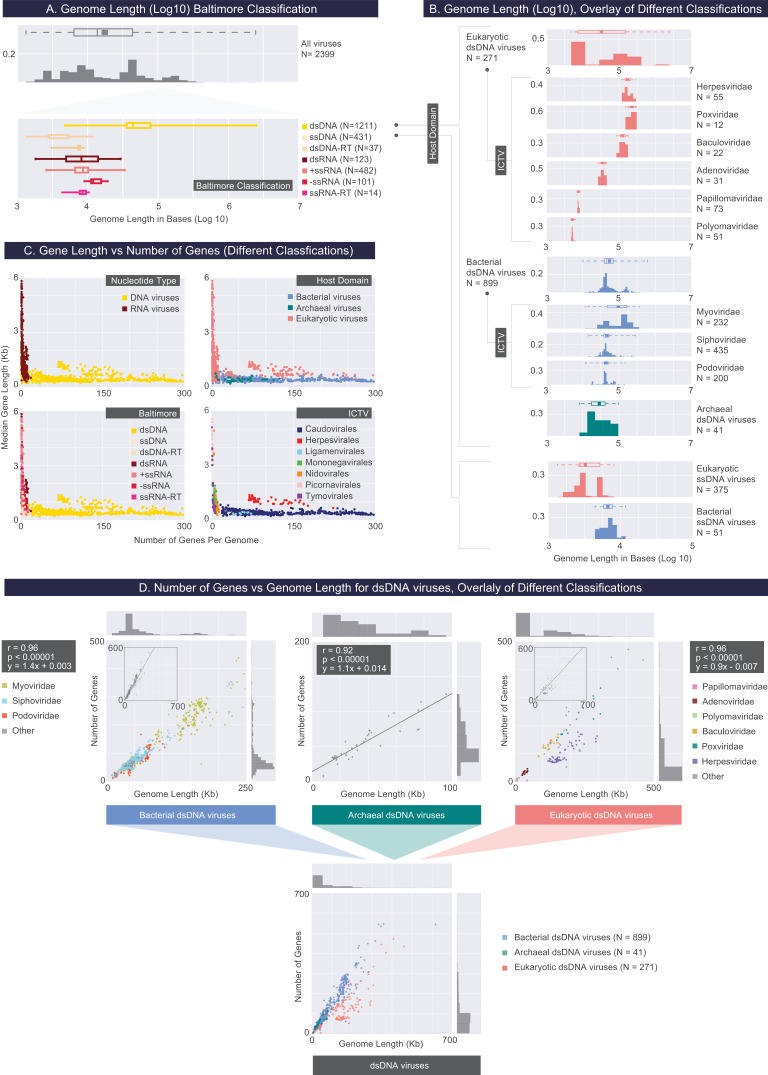
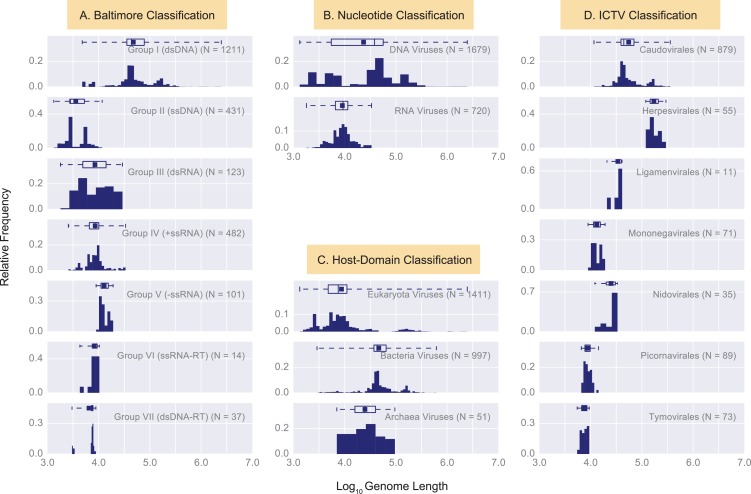
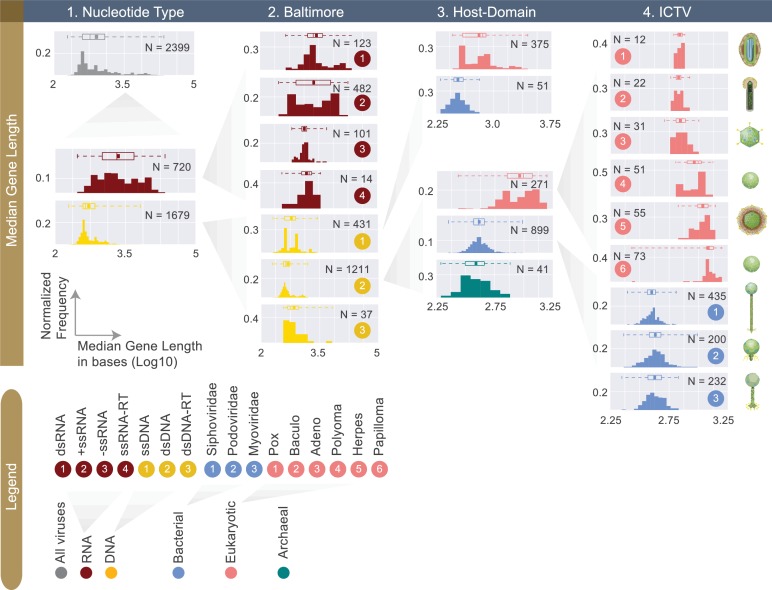
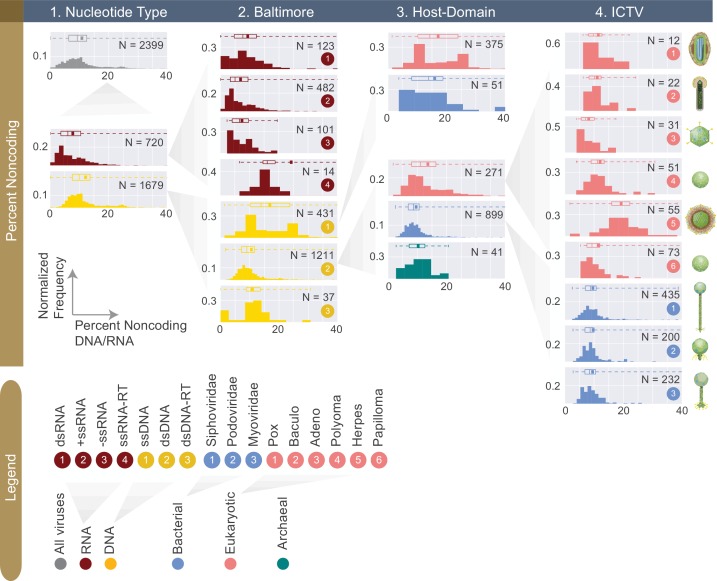
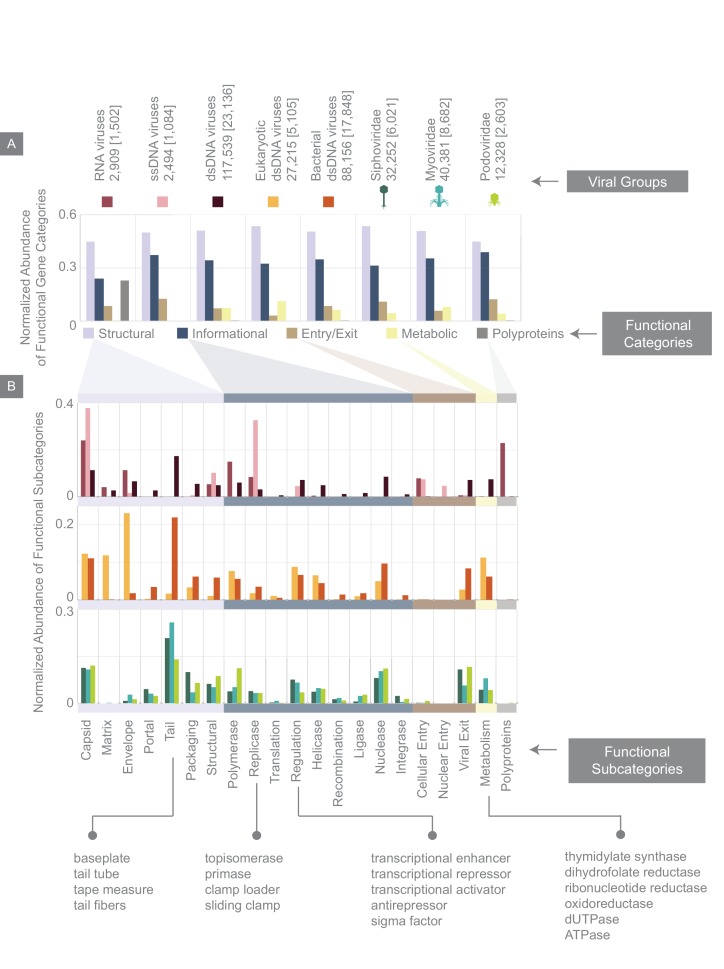
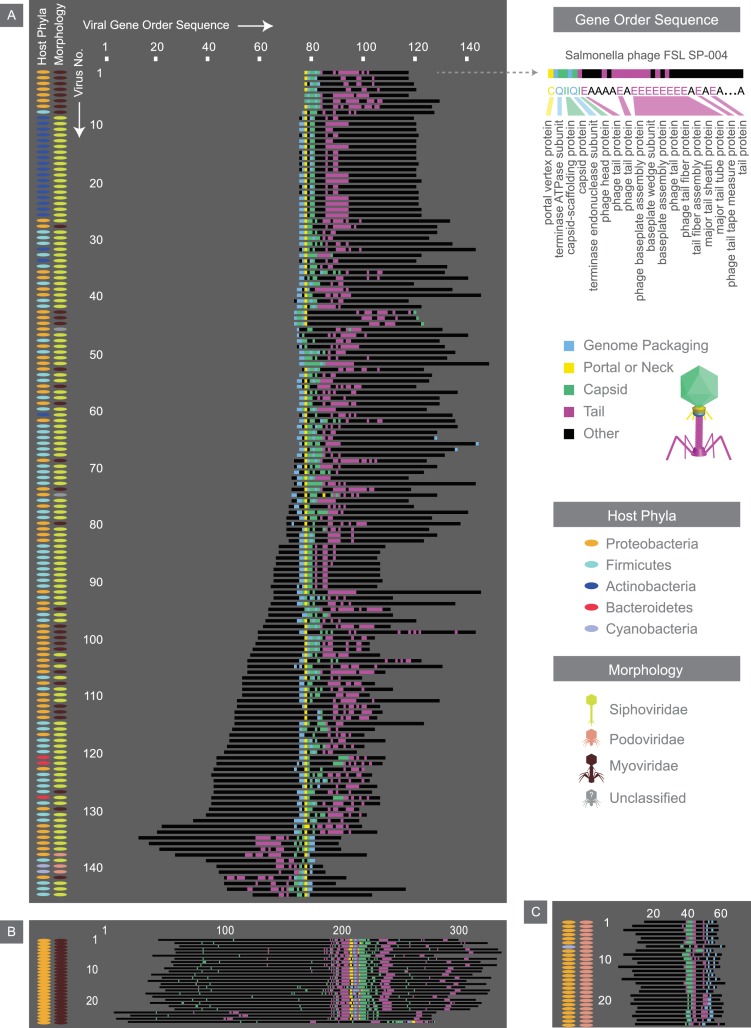
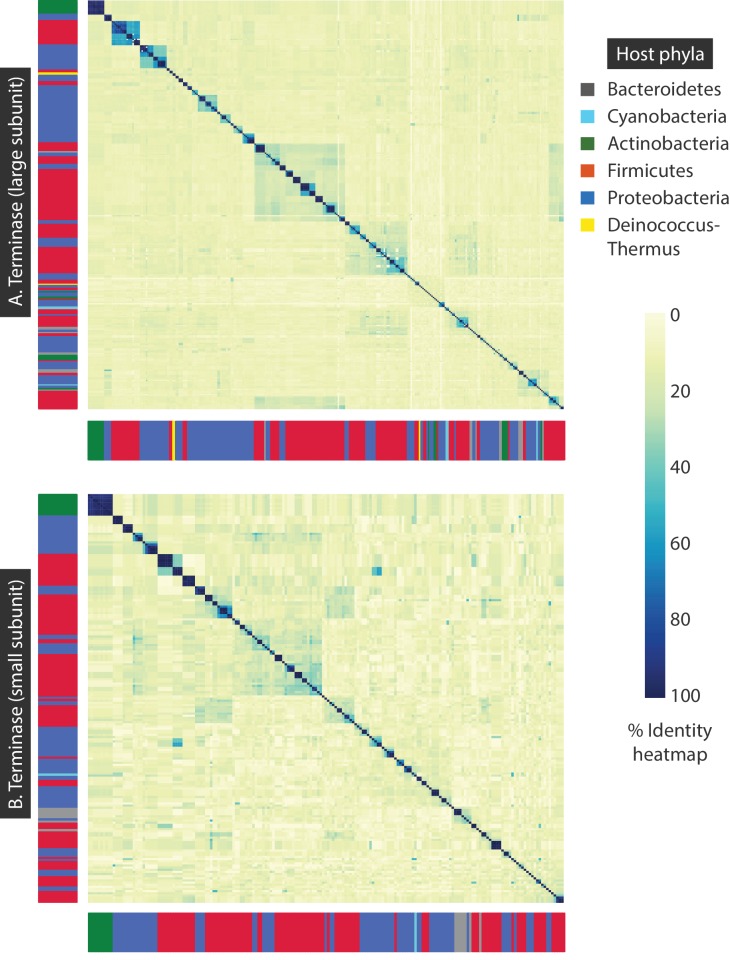
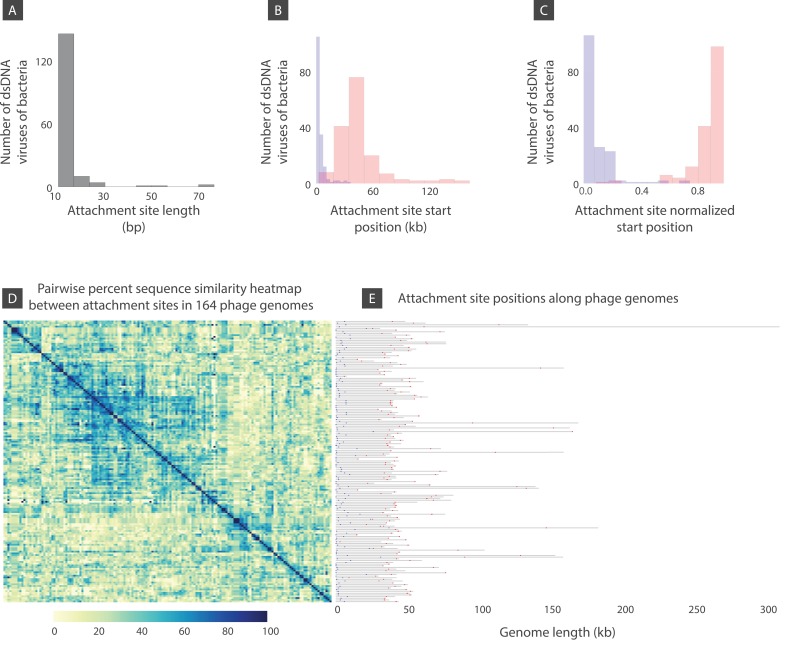
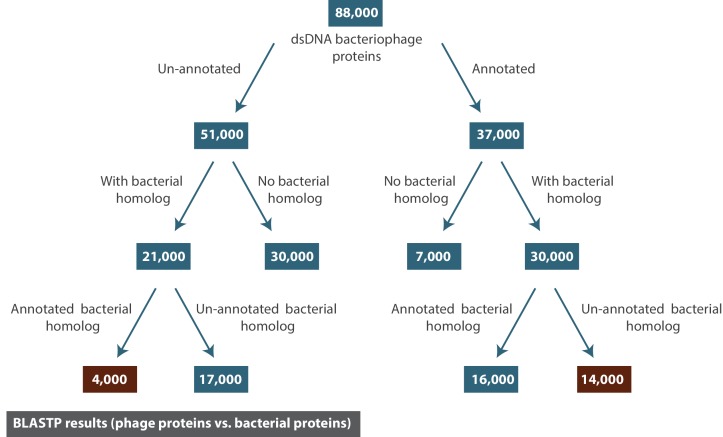
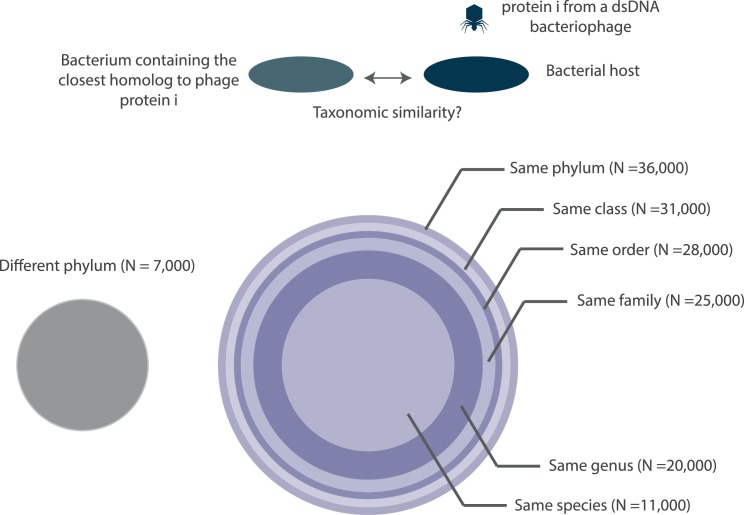
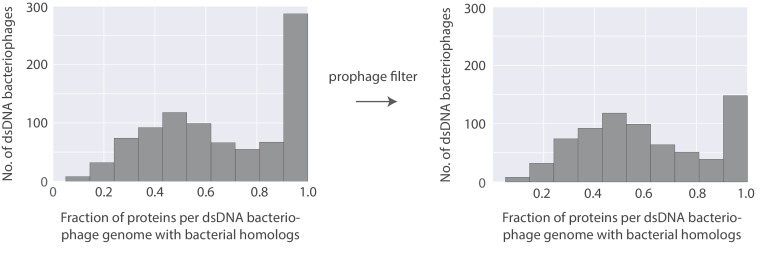
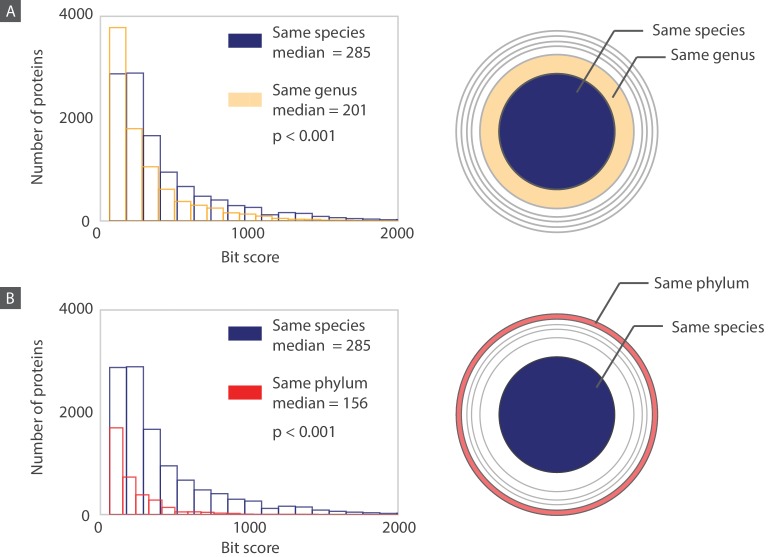
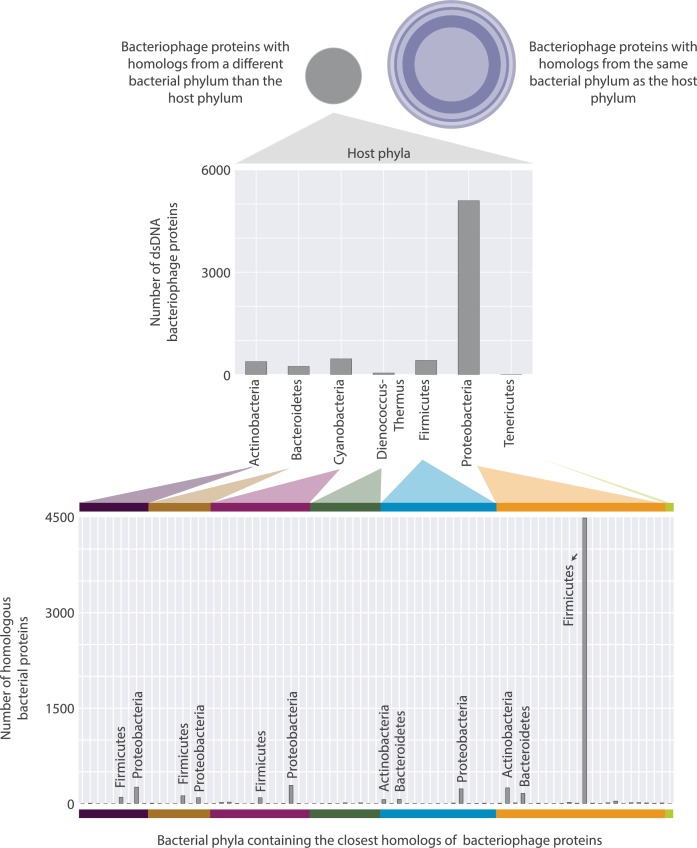
The complete assembly of viral genomes from metagenomic datasets (short genomic sequences gathered from environmental samples) has proven to be challenging, so there are significant blind spots when we view viral genomes through the lens of metagenomics. One approach to overcoming this problem is to leverage the thousands of complete viral genomes that are publicly available. Here we describe our efforts to assemble a comprehensive resource that provides a quantitative snapshot of viral genomic trends - such as gene density, noncoding percentage, and abundances of functional gene categories - across thousands of viral genomes. We have also developed a coarse-grained method for visualizing viral genome organization for hundreds of genomes at once, and have explored the extent of the overlap between bacterial and bacteriophage gene pools. Existing viral classification systems were developed prior to the sequencing era, so we present our analysis in a way that allows us to assess the utility of the different classification systems for capturing genomic trends.
Keywords: NCBI viral database; Viral classification; Viral genomic organization; Viral noncoding percentage; bacteriophages; infectious disease; microbiology; phage host interaction; virus.
© 2018, Mahmoudabadi et al.
Conflict of interest statement
GM, RP No competing interests declared
Figures
References
-
- Alberts B, Johnson A, Lewis J, Walter P, Raff M, Roberts K. Molecular Biology of the Cell 4th Edition: International Student Edition. Routledge; 2002.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
