

Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer
- PMID: 29625048
- PMCID: PMC5957518
- DOI: 10.1016/j.cell.2018.03.022
Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer
Abstract
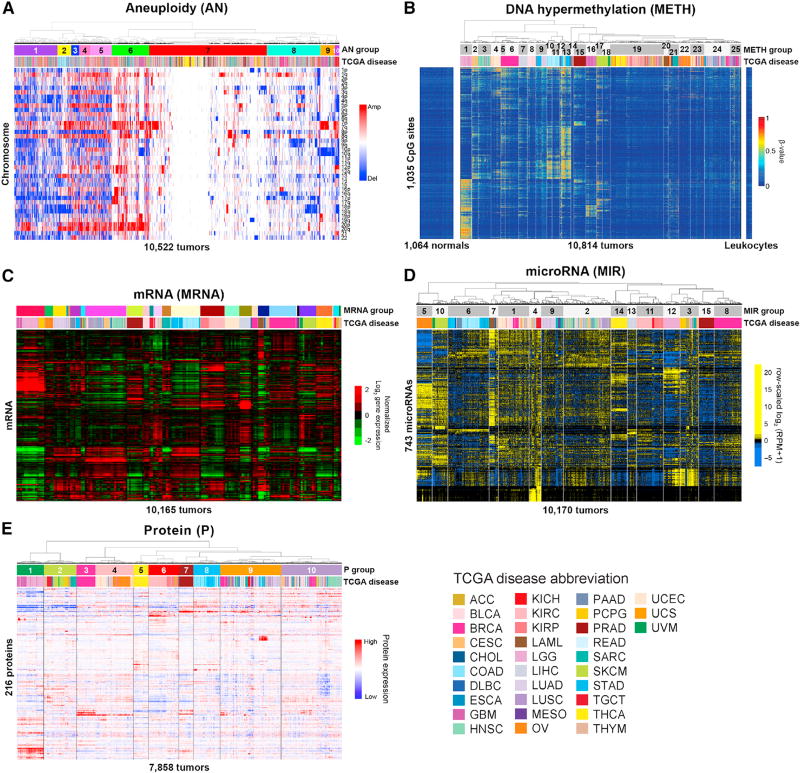
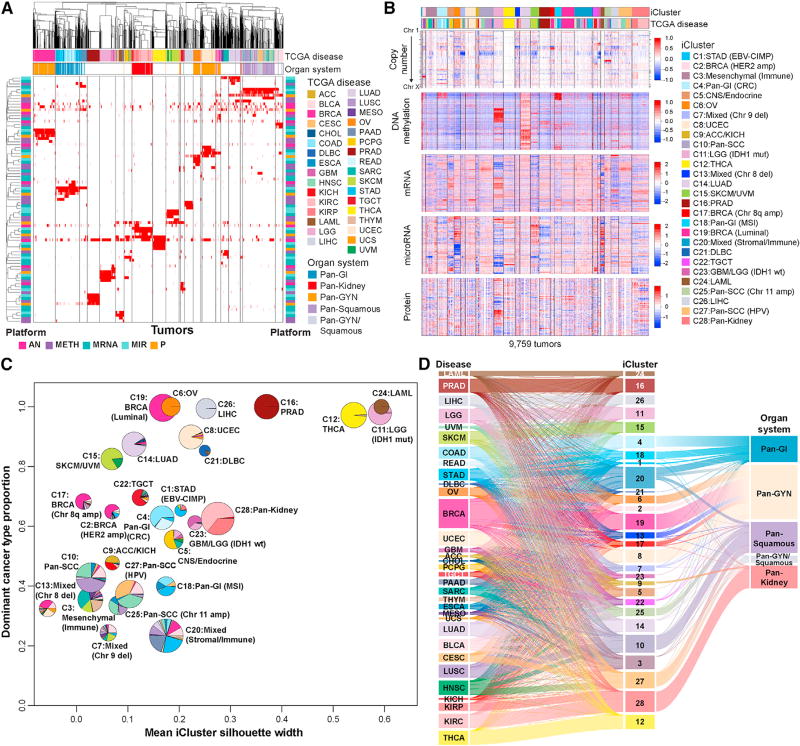
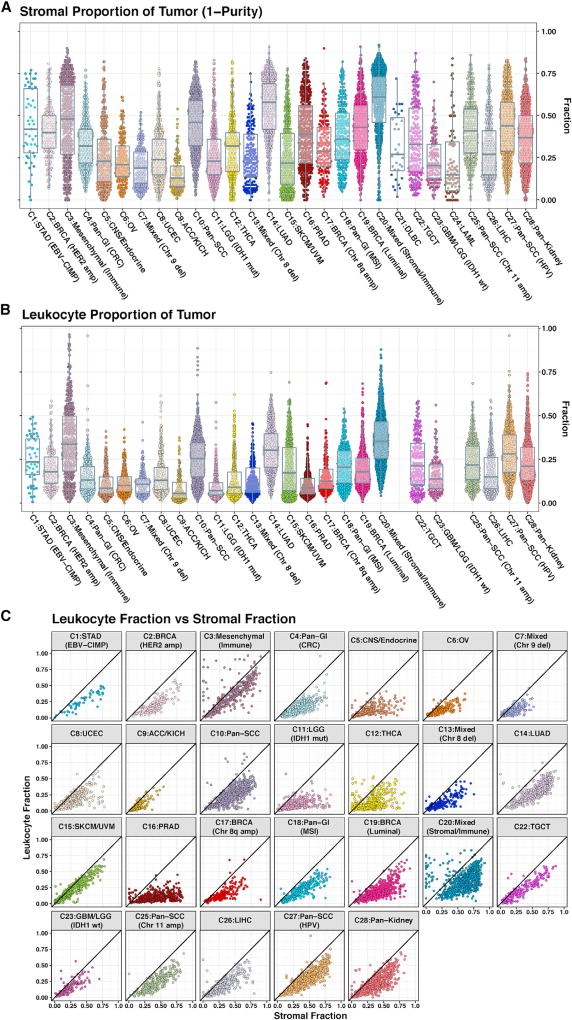
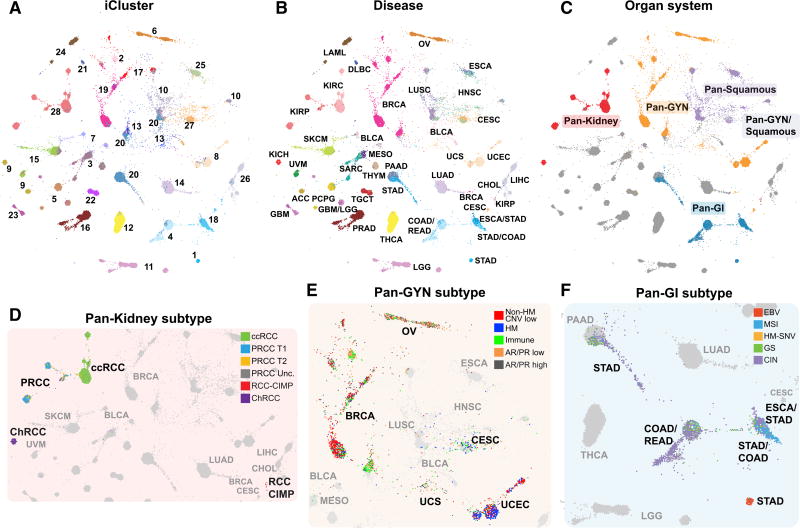
We conducted comprehensive integrative molecular analyses of the complete set of tumors in The Cancer Genome Atlas (TCGA), consisting of approximately 10,000 specimens and representing 33 types of cancer. We performed molecular clustering using data on chromosome-arm-level aneuploidy, DNA hypermethylation, mRNA, and miRNA expression levels and reverse-phase protein arrays, of which all, except for aneuploidy, revealed clustering primarily organized by histology, tissue type, or anatomic origin. The influence of cell type was evident in DNA-methylation-based clustering, even after excluding sites with known preexisting tissue-type-specific methylation. Integrative clustering further emphasized the dominant role of cell-of-origin patterns. Molecular similarities among histologically or anatomically related cancer types provide a basis for focused pan-cancer analyses, such as pan-gastrointestinal, pan-gynecological, pan-kidney, and pan-squamous cancers, and those related by stemness features, which in turn may inform strategies for future therapeutic development.
Keywords: TCGA; cancer; cell-of-origin; genome; methylome; organs; proteome; subtypes; tissues; transcriptome.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures

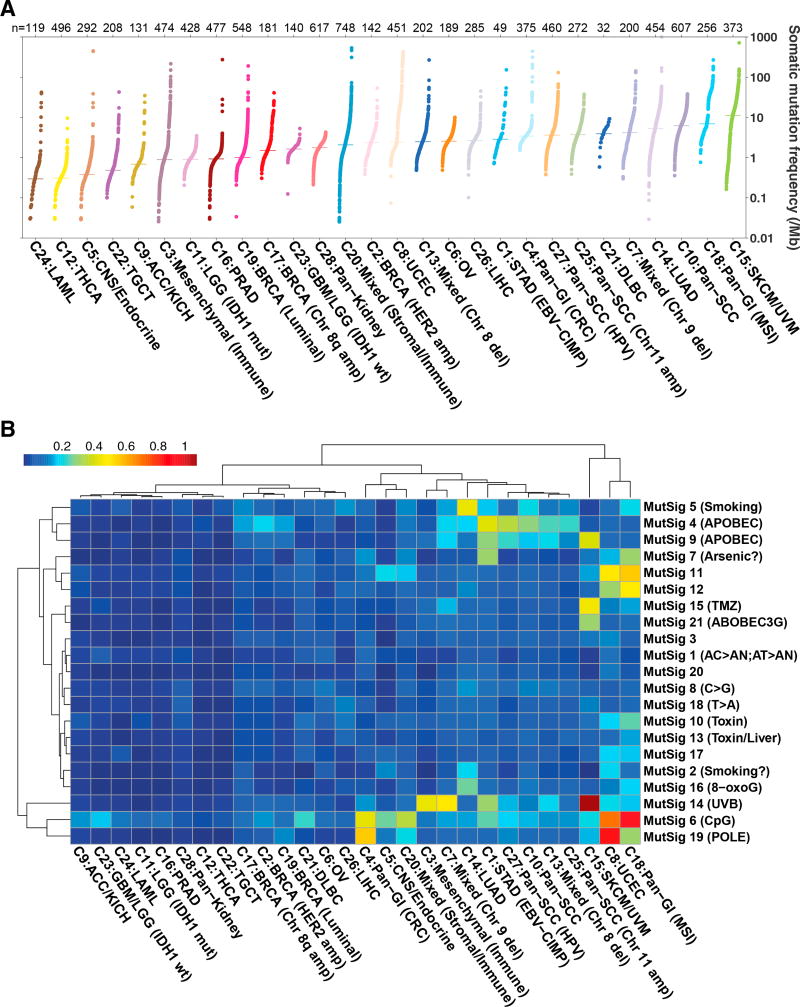
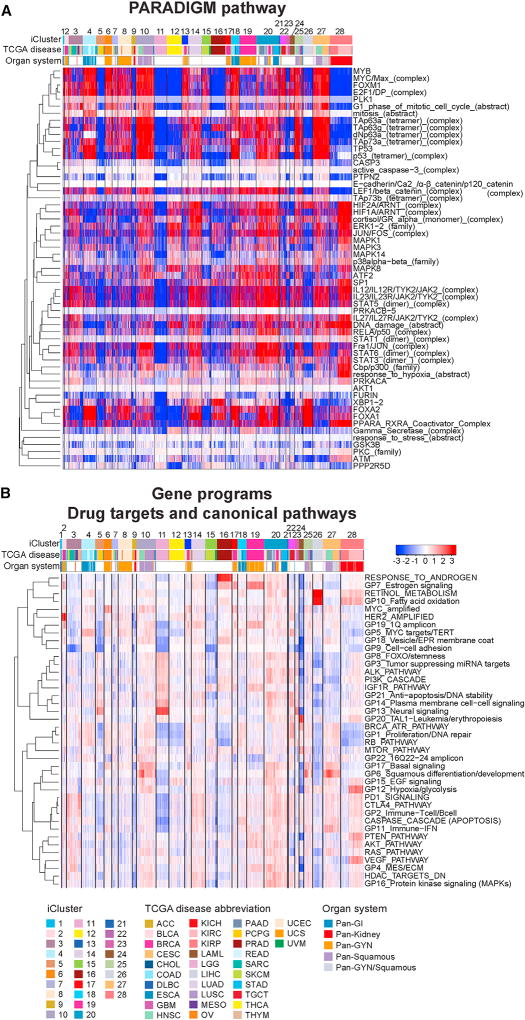
Comment in
-
A pan-cancer atlas.Nat Methods. 2018 Jun;15(6):407. doi: 10.1038/s41592-018-0020-4. Nat Methods. 2018. PMID: 29855579 No abstract available.
References
-
- Alencar A, Polley T. DrL (VxOrd) 2011 http://wiki.cns.iu.edu/pages/viewpage.action?pageId=1704113.
-
- Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, Liu Y, Fan H, Shen H, Ravikumar V, et al. A comprehensive Pan-Cancer molecular study of gynecologic and breast cancers. Cancer Cell. 2018;33 https://doi.org/10.1016/j.ccell.2018.03.014. - DOI - PMC - PubMed
-
- Calabrò A, Beissbarth T, Kuner R, Stojanov M, Benner A, Asslaber M, Ploner F, Zatloukal K, Samonigg H, Poustka A, Sültmann H. Effects of infiltrating lymphocytes and estrogen receptor on gene expression and prognosis in breast cancer. Breast Cancer Res. Treat. 2009;116:69–77. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U24 CA143882/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- P30 DK058404/DK/NIDDK NIH HHS/United States
- P30 CA016086/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- P50 CA058223/CA/NCI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24 CA210974/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U24 CA210949/CA/NCI NIH HHS/United States
- R01 CA163722/CA/NCI NIH HHS/United States
- R50 CA221675/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24 CA210969/CA/NCI NIH HHS/United States
- U24 CA210988/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- U24 CA199461/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
