

Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics
- PMID: 29625049
- PMCID: PMC5916814
- DOI: 10.1016/j.cell.2018.03.033
Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics
Abstract
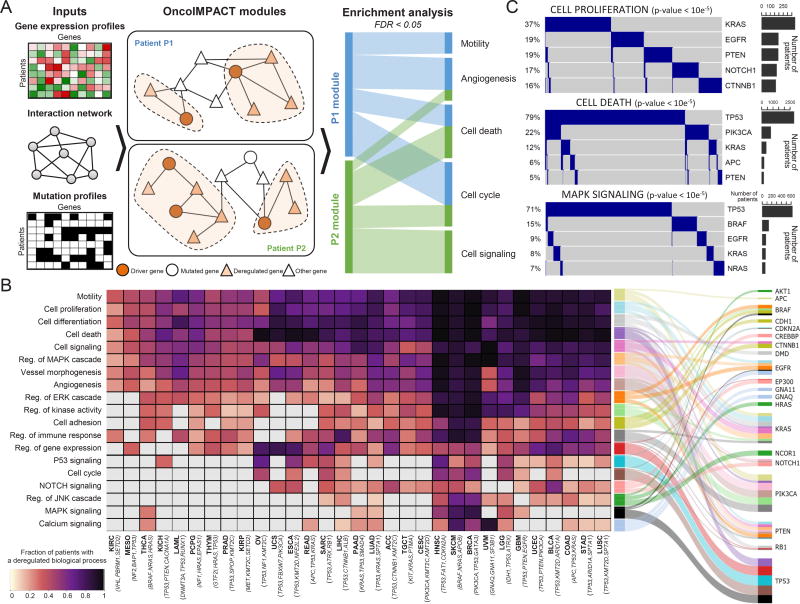
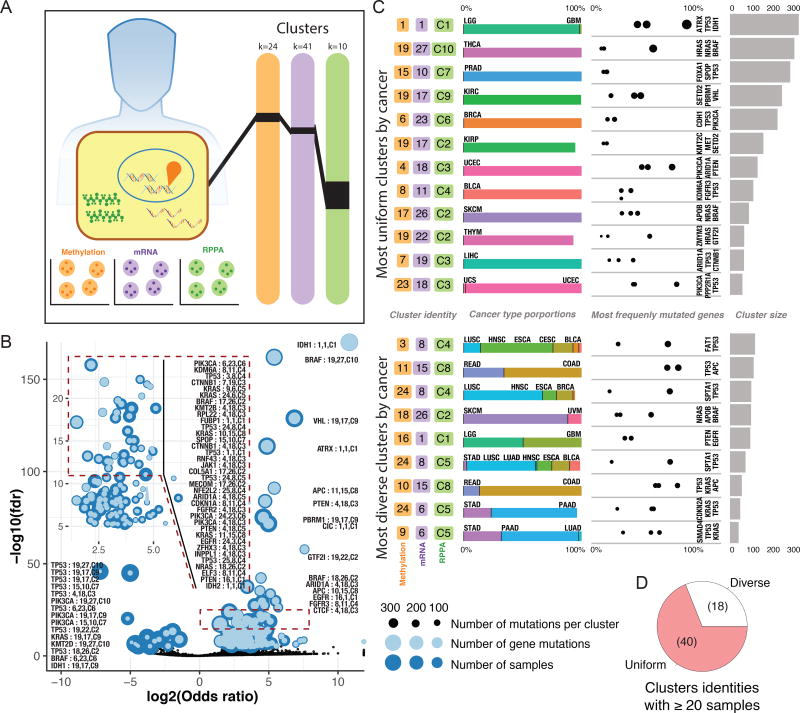
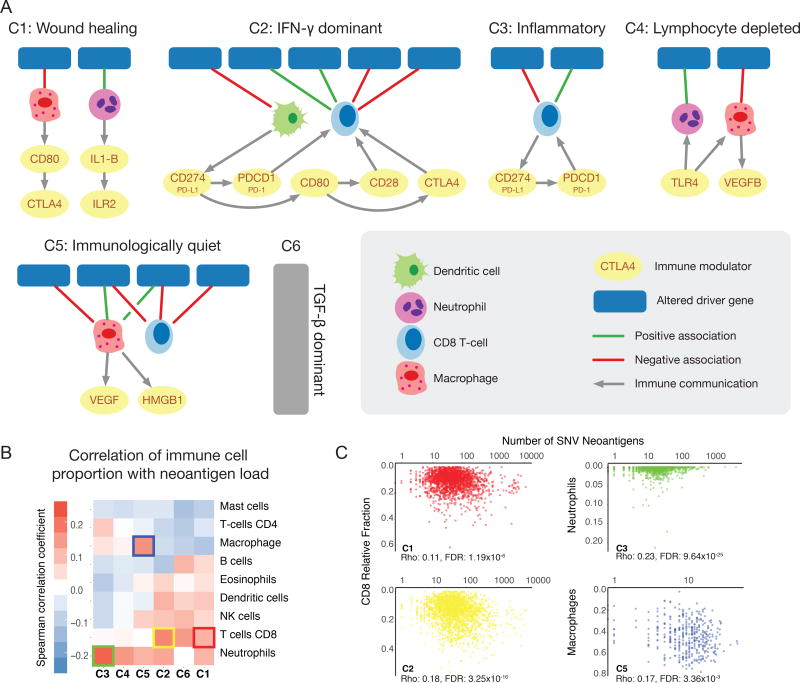
The Cancer Genome Atlas (TCGA) has catalyzed systematic characterization of diverse genomic alterations underlying human cancers. At this historic junction marking the completion of genomic characterization of over 11,000 tumors from 33 cancer types, we present our current understanding of the molecular processes governing oncogenesis. We illustrate our insights into cancer through synthesis of the findings of the TCGA PanCancer Atlas project on three facets of oncogenesis: (1) somatic driver mutations, germline pathogenic variants, and their interactions in the tumor; (2) the influence of the tumor genome and epigenome on transcriptome and proteome; and (3) the relationship between tumor and the microenvironment, including implications for drugs targeting driver events and immunotherapies. These results will anchor future characterization of rare and common tumor types, primary and relapsed tumors, and cancers across ancestry groups and will guide the deployment of clinical genomic sequencing.
Keywords: TCGA; cancer; cancer genomics; omics; oncogenic process.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Bassi R, Giussani P, Anelli V, Colleoni T, Pedrazzi M, Patrone M, Viani P, Sparatore B, Melloni E, Riboni L. HMGB1 as an autocrine stimulus in human T98G glioblastoma cells: role in cell growth and migration. J Neurooncol. 2008;87:23–33. - PubMed
Publication types
MeSH terms
Grants and funding
- P30 ES010126/ES/NIEHS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- U24 CA143882/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- P30 CA016086/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24 CA210974/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24 CA210949/CA/NCI NIH HHS/United States
- U24 CA210988/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA211006/CA/NCI NIH HHS/United States
- R01 CA163722/CA/NCI NIH HHS/United States
- R25 DA027995/DA/NIDA NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- R50 CA221675/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
