

Pathogenic Germline Variants in 10,389 Adult Cancers
- PMID: 29625052
- PMCID: PMC5949147
- DOI: 10.1016/j.cell.2018.03.039
Pathogenic Germline Variants in 10,389 Adult Cancers
Abstract
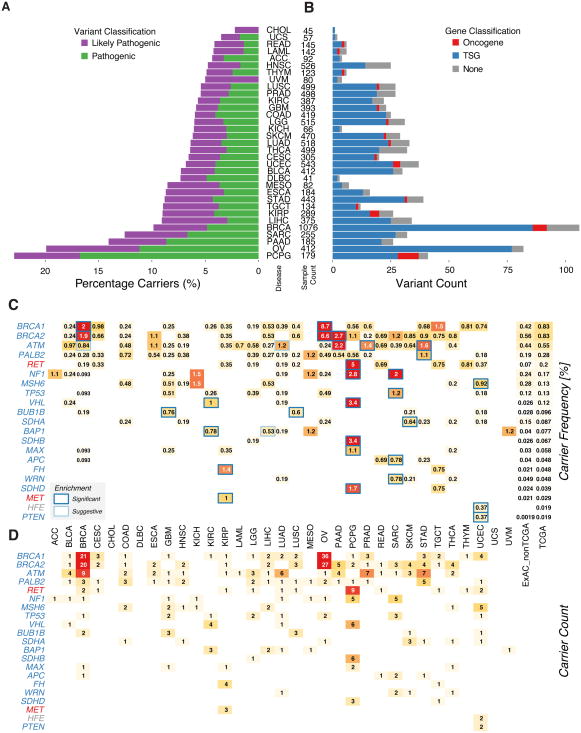
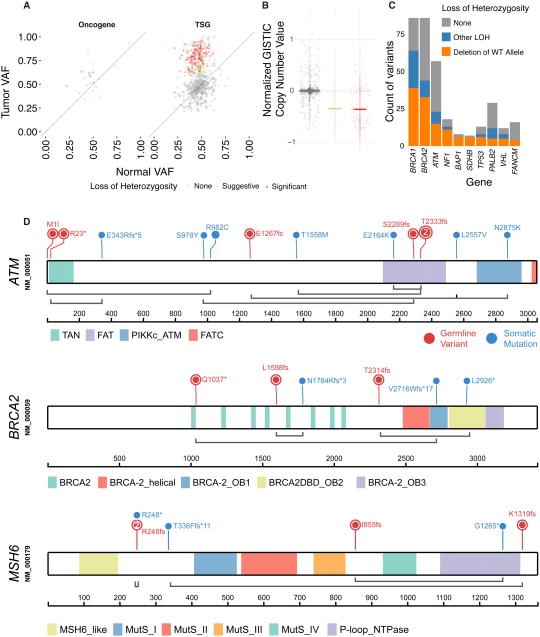
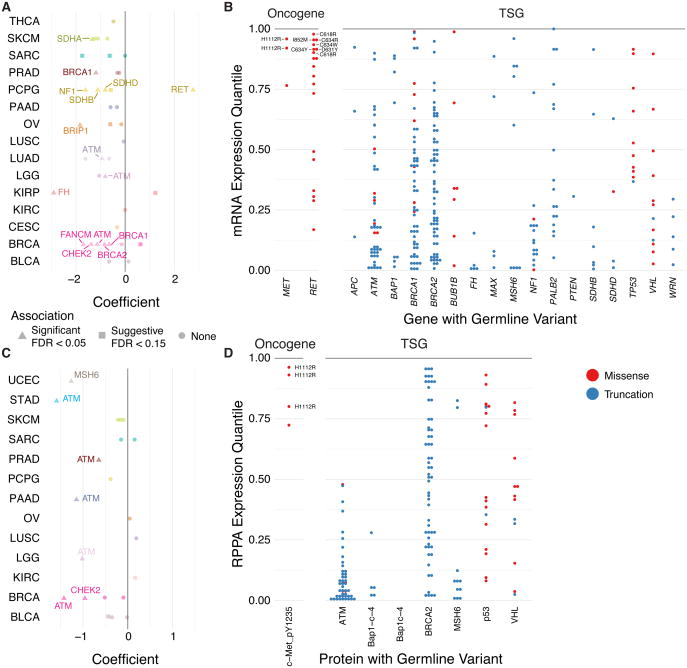
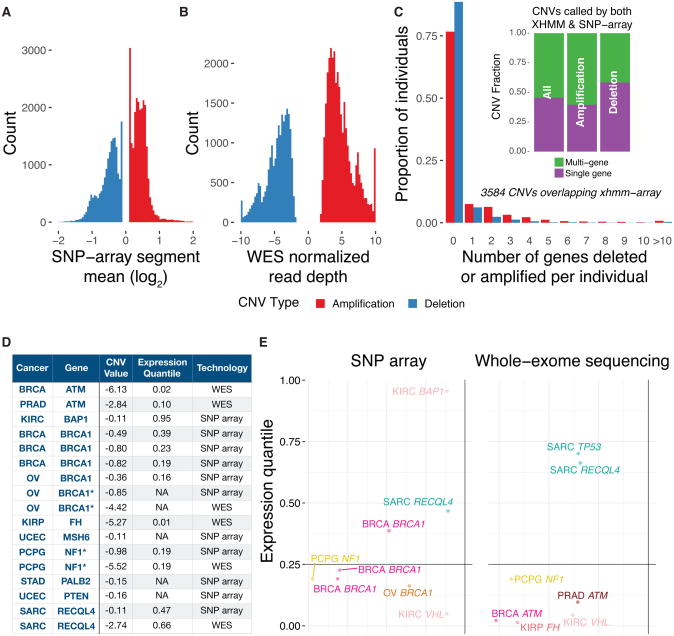
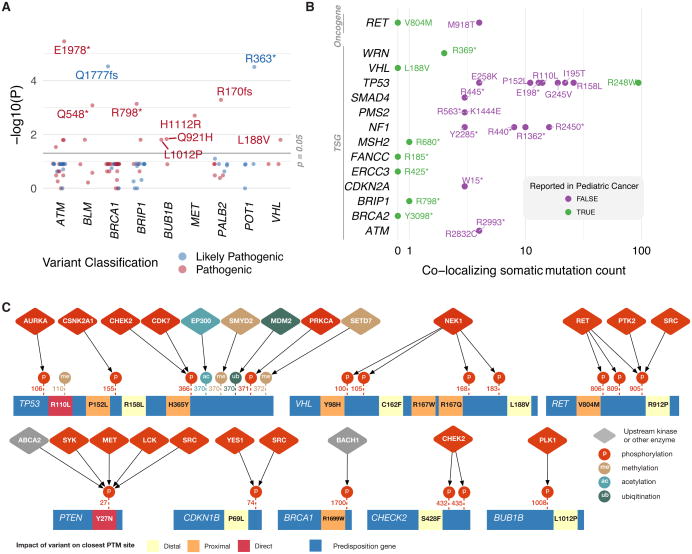
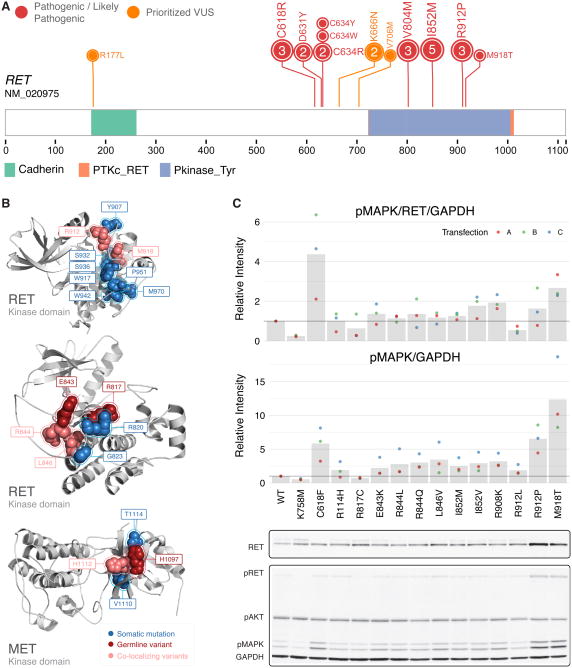
We conducted the largest investigation of predisposition variants in cancer to date, discovering 853 pathogenic or likely pathogenic variants in 8% of 10,389 cases from 33 cancer types. Twenty-one genes showed single or cross-cancer associations, including novel associations of SDHA in melanoma and PALB2 in stomach adenocarcinoma. The 659 predisposition variants and 18 additional large deletions in tumor suppressors, including ATM, BRCA1, and NF1, showed low gene expression and frequent (43%) loss of heterozygosity or biallelic two-hit events. We also discovered 33 such variants in oncogenes, including missenses in MET, RET, and PTPN11 associated with high gene expression. We nominated 47 additional predisposition variants from prioritized VUSs supported by multiple evidences involving case-control frequency, loss of heterozygosity, expression effect, and co-localization with mutations and modified residues. Our integrative approach links rare predisposition variants to functional consequences, informing future guidelines of variant classification and germline genetic testing in cancer.
Keywords: LOH; cancer predisposition; germline and somatic genomes; variant pathogenicity.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, Berg JS, Biswas S, Bowling KM, Conlin LK, et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98:1067–1076. - PMC - PubMed
-
- Ballinger ML, Goode DL, Ray-Coquard I, James PA, Mitchell G, Niedermayr E, Puri A, Schiffman JD, Dite GS, Cipponi A, et al. Monogenic and polygenic determinants of sarcoma risk: an international genetic study. Lancet Oncol. 2016;17:1261–1271. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U24 CA143882/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24 CA210972/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- U24 CA210949/CA/NCI NIH HHS/United States
- R01 CA163722/CA/NCI NIH HHS/United States
- R01 CA178383/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- U24 CA211000/CA/NCI NIH HHS/United States
- R01 GM079656/GM/NIGMS NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- R01 DK102520/DK/NIDDK NIH HHS/United States
- K12 GM084897/GM/NIGMS NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- R01 CA180006/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24 CA210969/CA/NCI NIH HHS/United States
- U24 CA210988/CA/NCI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA211006/CA/NCI NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- R01 HG009711/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
