

Comprehensive Characterization of Cancer Driver Genes and Mutations
- PMID: 29625053
- PMCID: PMC6029450
- DOI: 10.1016/j.cell.2018.02.060
Comprehensive Characterization of Cancer Driver Genes and Mutations
Erratum in
-
Comprehensive Characterization of Cancer Driver Genes and Mutations.Cell. 2018 Aug 9;174(4):1034-1035. doi: 10.1016/j.cell.2018.07.034. Cell. 2018. PMID: 30096302 Free PMC article. No abstract available.
Abstract
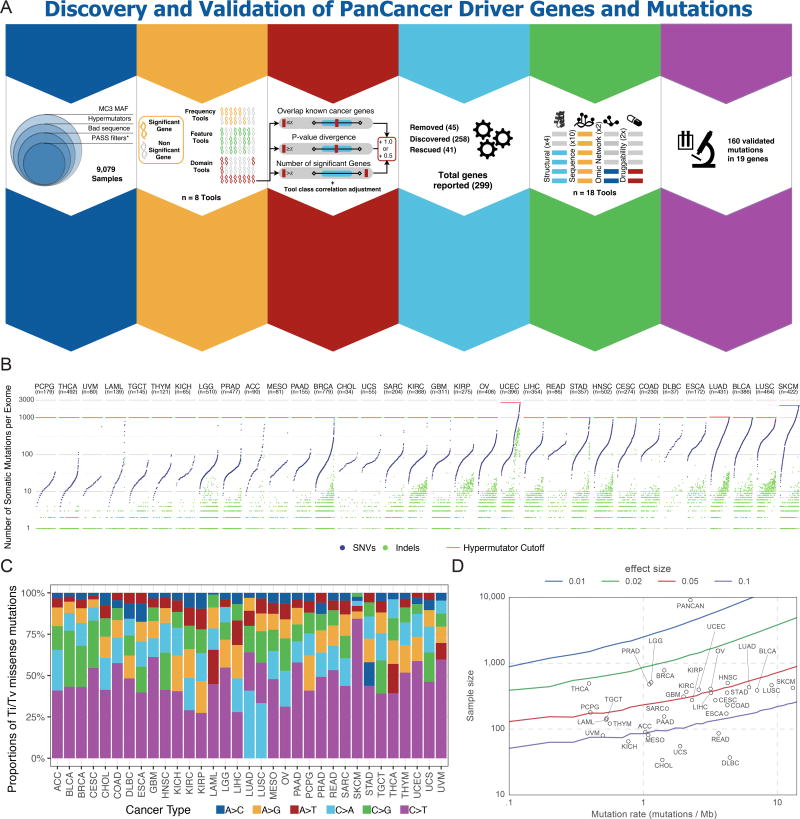
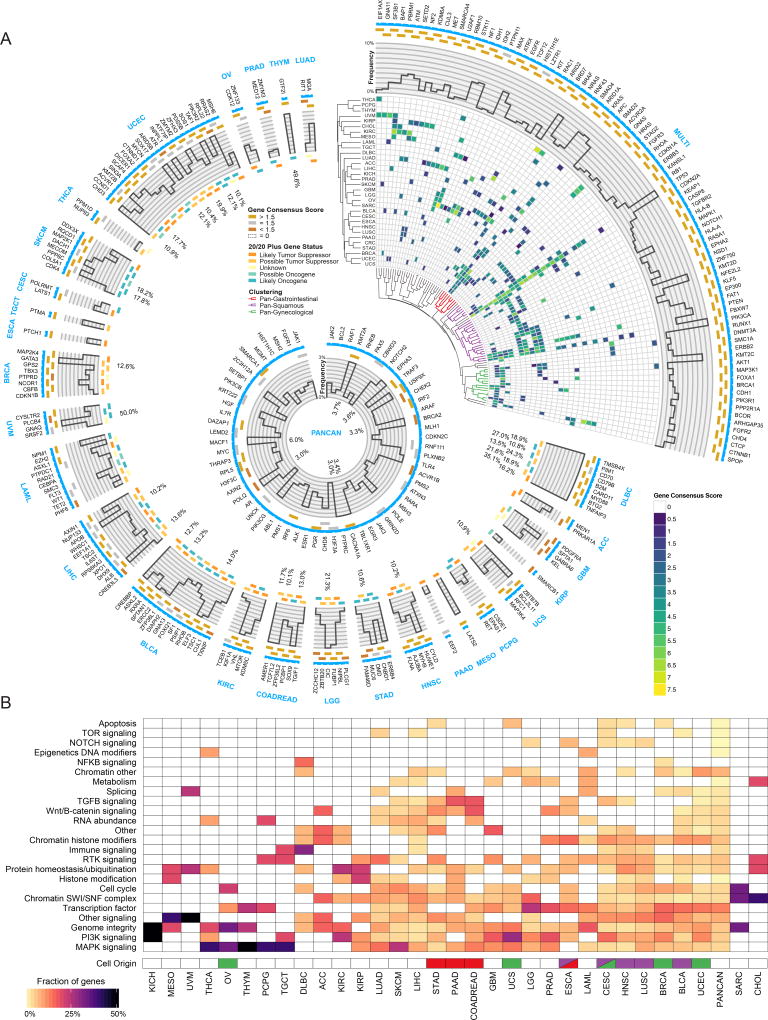
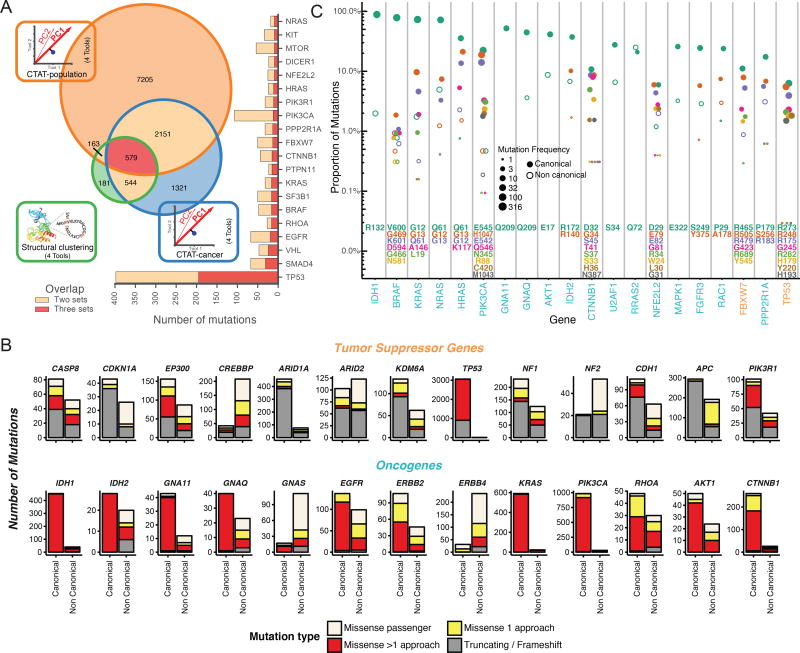
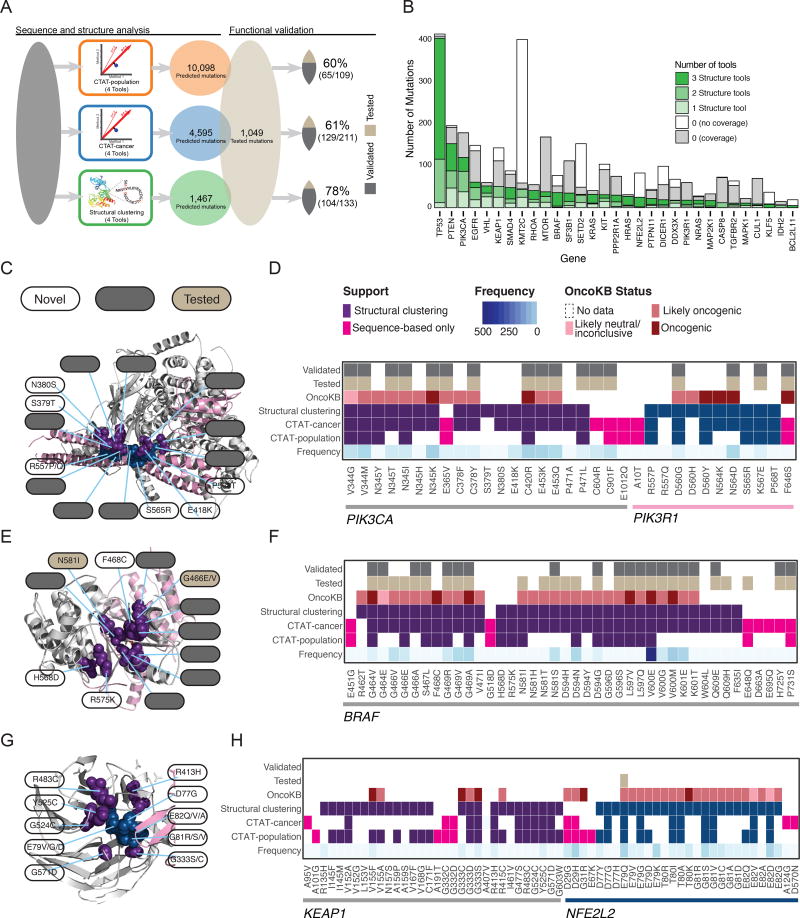
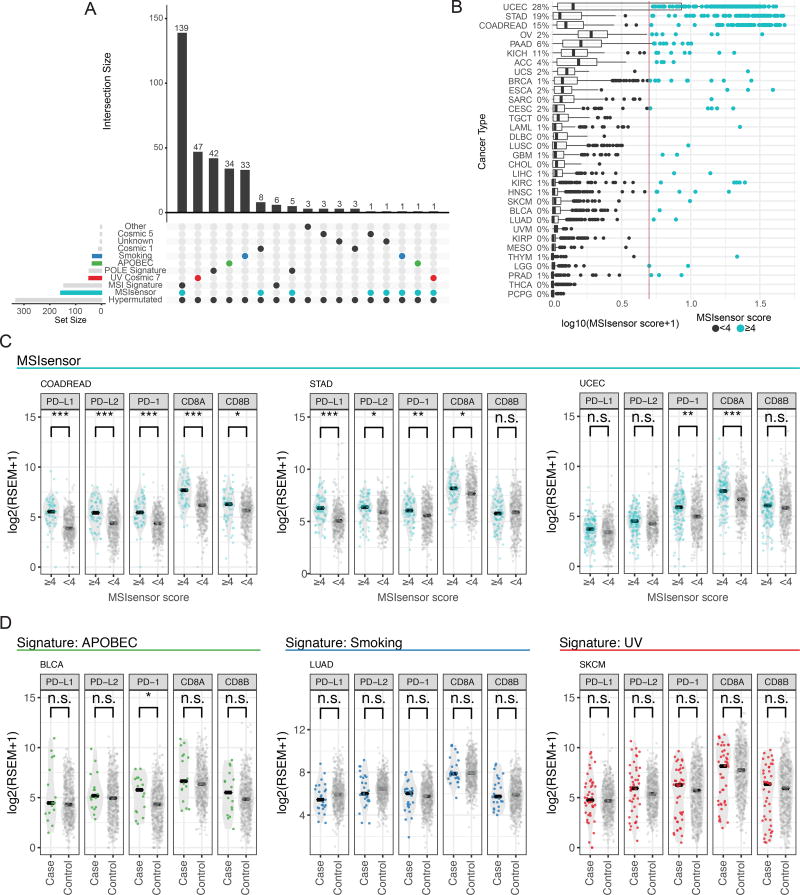
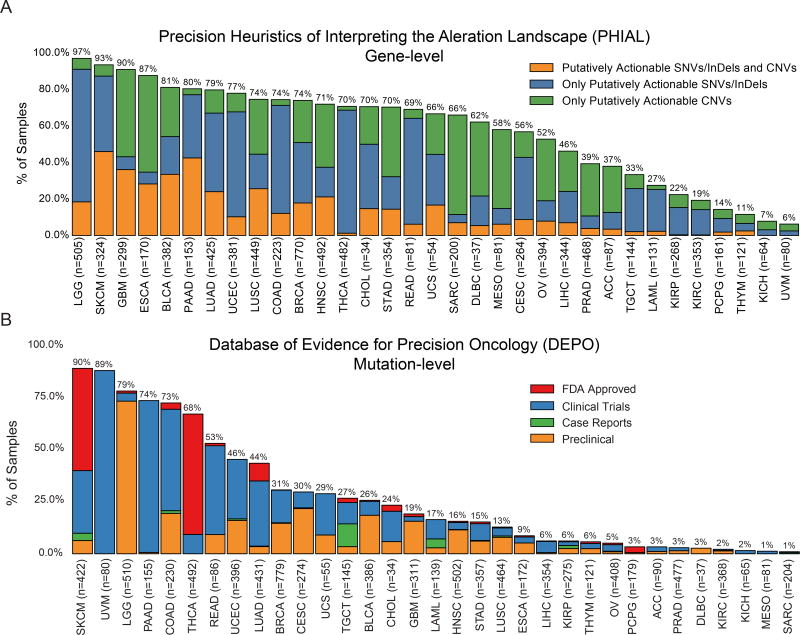
Identifying molecular cancer drivers is critical for precision oncology. Multiple advanced algorithms to identify drivers now exist, but systematic attempts to combine and optimize them on large datasets are few. We report a PanCancer and PanSoftware analysis spanning 9,423 tumor exomes (comprising all 33 of The Cancer Genome Atlas projects) and using 26 computational tools to catalog driver genes and mutations. We identify 299 driver genes with implications regarding their anatomical sites and cancer/cell types. Sequence- and structure-based analyses identified >3,400 putative missense driver mutations supported by multiple lines of evidence. Experimental validation confirmed 60%-85% of predicted mutations as likely drivers. We found that >300 MSI tumors are associated with high PD-1/PD-L1, and 57% of tumors analyzed harbor putative clinically actionable events. Our study represents the most comprehensive discovery of cancer genes and mutations to date and will serve as a blueprint for future biological and clinical endeavors.
Keywords: driver gene discovery; mutations of clinical relevance; oncology; structure analysis.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests.
Figures
References
-
- Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. Journal of clinical oncology. 2008;26:2139–2146. - PMC - PubMed
-
- Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. The lancet oncology. 2013;14:249–256. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U24 CA143882/CA/NCI NIH HHS/United States
- U24 CA143866/CA/NCI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- U24 CA143843/CA/NCI NIH HHS/United States
- U24 CA143848/CA/NCI NIH HHS/United States
- R01 CA178383/CA/NCI NIH HHS/United States
- U24 CA204817/CA/NCI NIH HHS/United States
- U24 CA210990/CA/NCI NIH HHS/United States
- F32 GM072403/GM/NIGMS NIH HHS/United States
- P30 CA016672/CA/NCI NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U24 CA143835/CA/NCI NIH HHS/United States
- U24 CA210950/CA/NCI NIH HHS/United States
- R21 CA135877/CA/NCI NIH HHS/United States
- U24 CA143845/CA/NCI NIH HHS/United States
- U24 CA143799/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- U24 CA144025/CA/NCI NIH HHS/United States
- S10 OD016290/OD/NIH HHS/United States
- U24 CA143840/CA/NCI NIH HHS/United States
- R01 CA180006/CA/NCI NIH HHS/United States
- U24 CA143858/CA/NCI NIH HHS/United States
- R21 CA152432/CA/NCI NIH HHS/United States
- U24 CA210957/CA/NCI NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- U24 CA143883/CA/NCI NIH HHS/United States
- U24 CA211006/CA/NCI NIH HHS/United States
- U24 CA210999/CA/NCI NIH HHS/United States
- R01 CA163722/CA/NCI NIH HHS/United States
- R25 DA027995/DA/NIDA NIH HHS/United States
- U24 CA143867/CA/NCI NIH HHS/United States
- R01 HG009711/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
