

A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples
- PMID: 29625054
- PMCID: PMC5890960
- DOI: 10.1016/j.cell.2018.03.027
A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples
Abstract
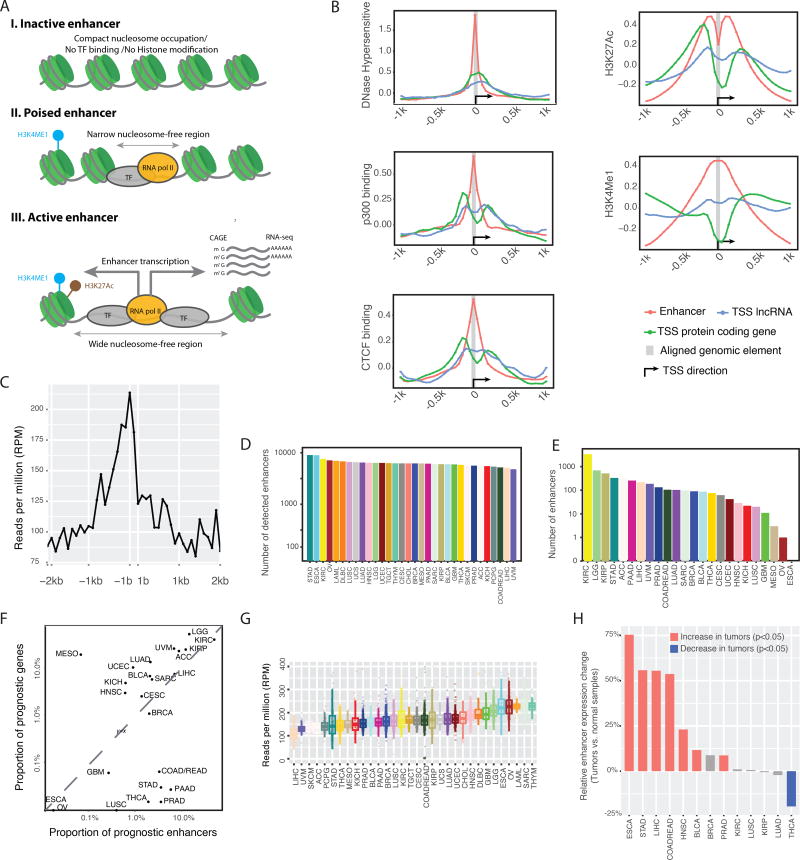
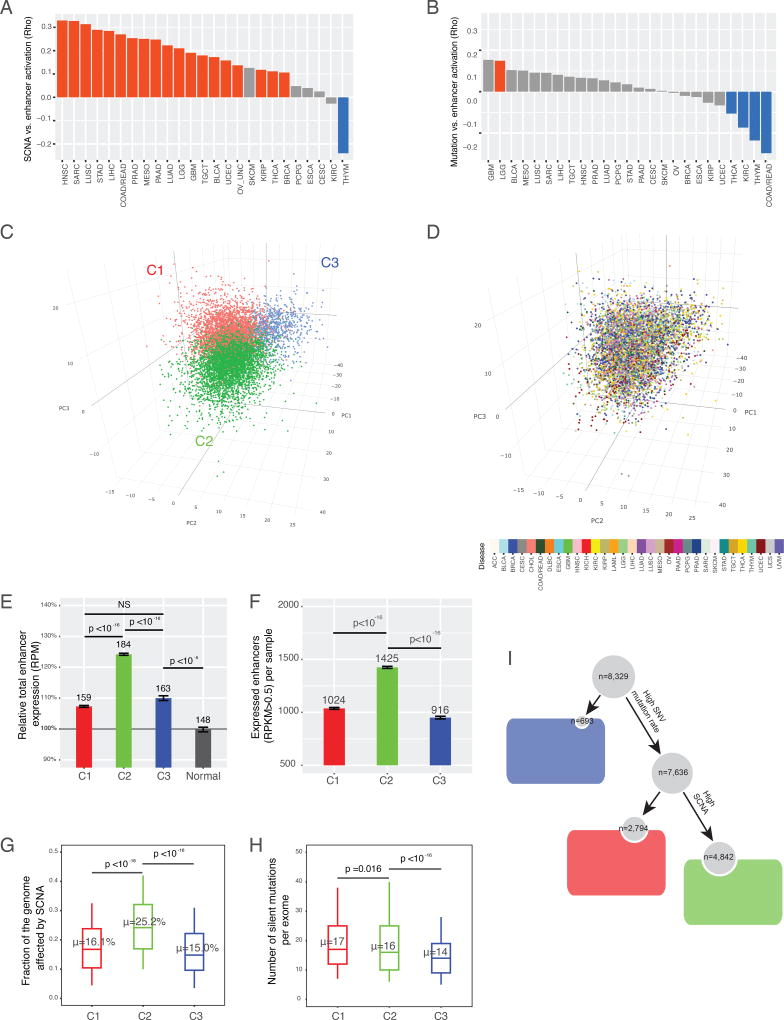
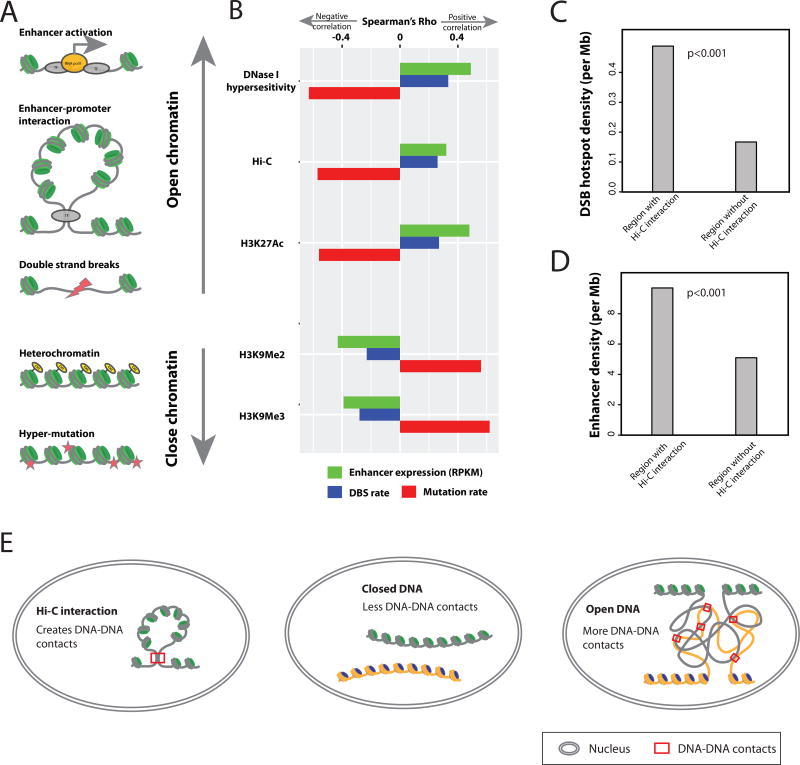
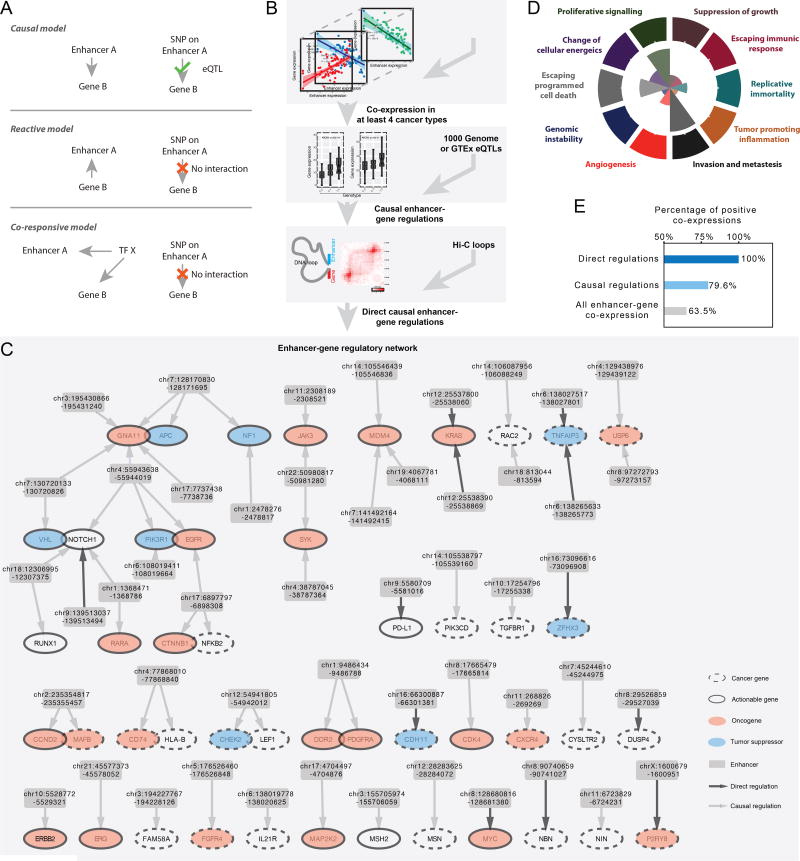
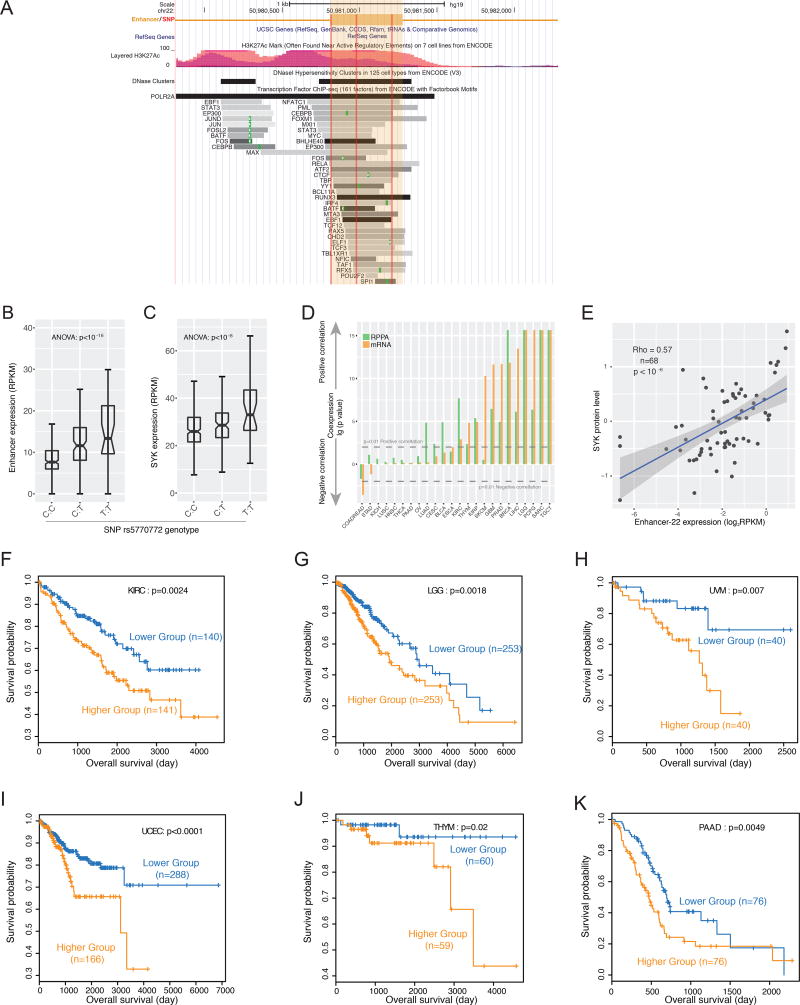
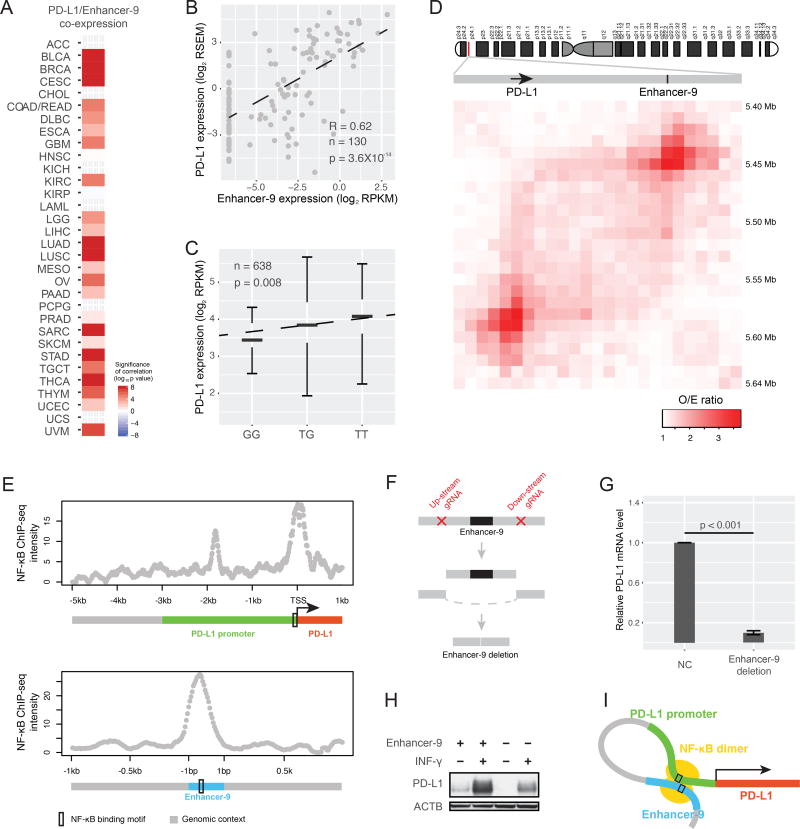
The role of enhancers, a key class of non-coding regulatory DNA elements, in cancer development has increasingly been appreciated. Here, we present the detection and characterization of a large number of expressed enhancers in a genome-wide analysis of 8928 tumor samples across 33 cancer types using TCGA RNA-seq data. Compared with matched normal tissues, global enhancer activation was observed in most cancers. Across cancer types, global enhancer activity was positively associated with aneuploidy, but not mutation load, suggesting a hypothesis centered on "chromatin-state" to explain their interplay. Integrating eQTL, mRNA co-expression, and Hi-C data analysis, we developed a computational method to infer causal enhancer-gene interactions, revealing enhancers of clinically actionable genes. Having identified an enhancer ∼140 kb downstream of PD-L1, a major immunotherapy target, we validated it experimentally. This study provides a systematic view of enhancer activity in diverse tumor contexts and suggests the clinical implications of enhancers.
Keywords: PD-L1 expression; The Cancer Genome Atlas; aneuploidy; chromatin state; enhancer expression; mutation burden; pan-cancer analysis; prognostic markers.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Aran D, Hellman A. DNA methylation of transcriptional enhancers and cancer predisposition. Cell. 2013;154:11–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
