

Lipid binding promotes the open conformation and tumor-suppressive activity of neurofibromin 2
- PMID: 29626191
- PMCID: PMC5889391
- DOI: 10.1038/s41467-018-03648-4
Lipid binding promotes the open conformation and tumor-suppressive activity of neurofibromin 2
Abstract
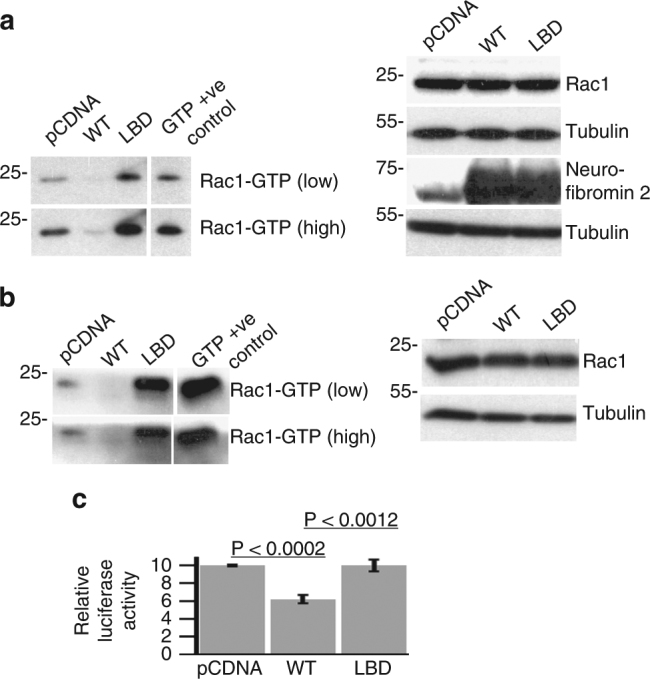
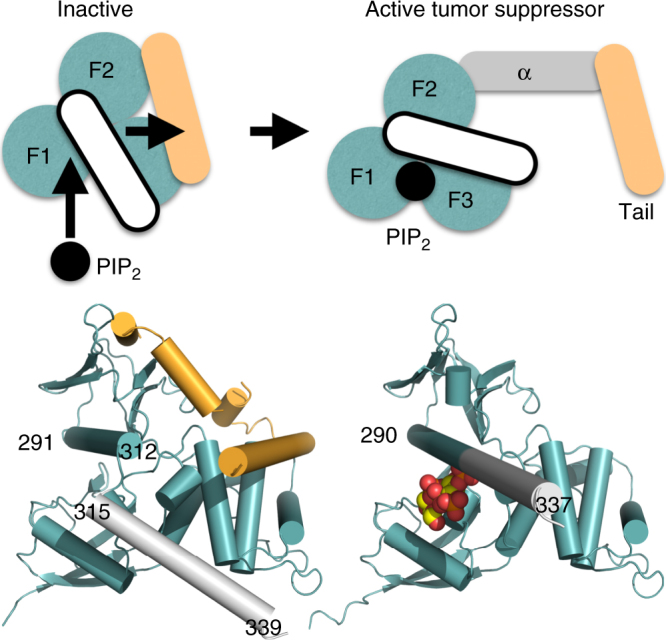
Neurofibromatosis type 2 (NF2) is a tumor-forming disease of the nervous system caused by deletion or by loss-of-function mutations in NF2, encoding the tumor suppressing protein neurofibromin 2 (also known as schwannomin or merlin). Neurofibromin 2 is a member of the ezrin, radixin, moesin (ERM) family of proteins regulating the cytoskeleton and cell signaling. The correlation of the tumor-suppressive function and conformation (open or closed) of neurofibromin 2 has been subject to much speculation, often based on extrapolation from other ERM proteins, and controversy. Here we show that lipid binding results in the open conformation of neurofibromin 2 and that lipid binding is necessary for inhibiting cell proliferation. Collectively, our results provide a mechanism in which the open conformation is unambiguously correlated with lipid binding and localization to the membrane, which are critical for the tumor-suppressive function of neurofibromin 2, thus finally reconciling the long-standing conformation and function debate.
Conflict of interest statement
The authors declare no competing interests.
Figures
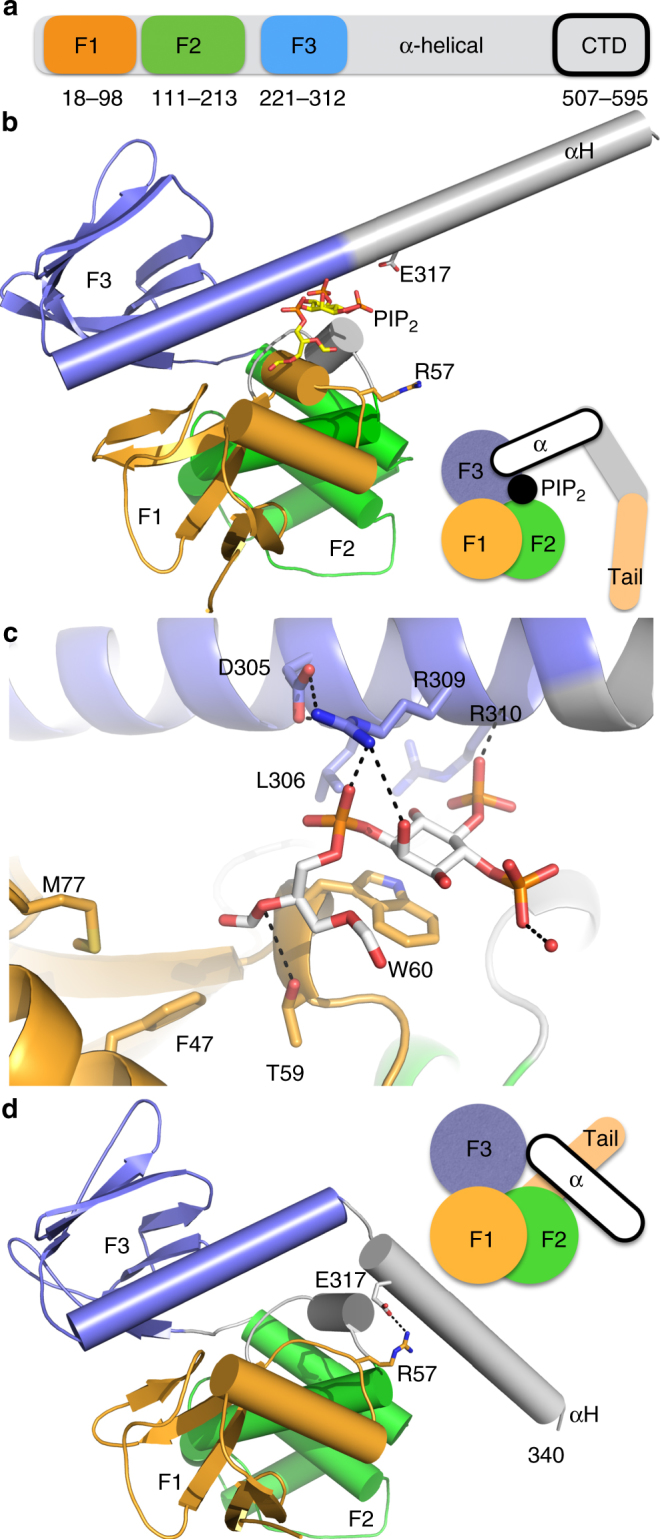
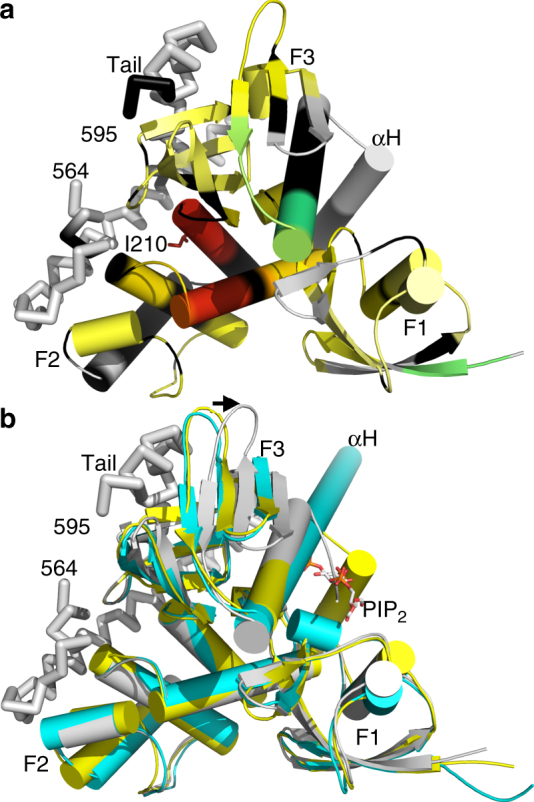
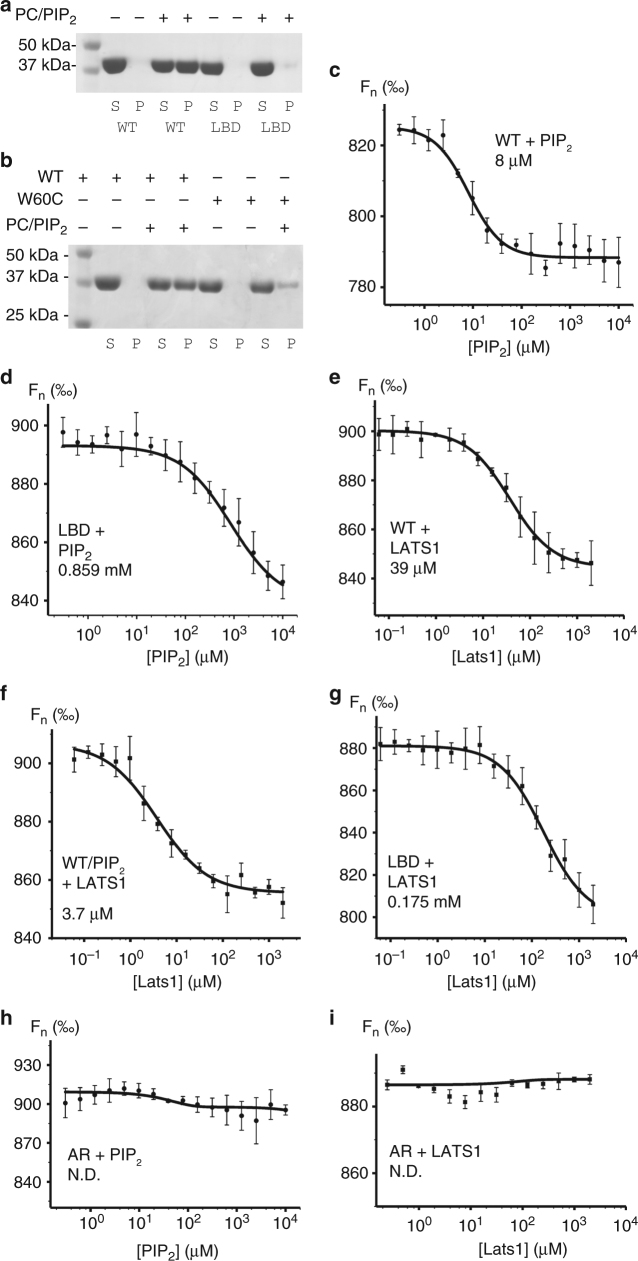
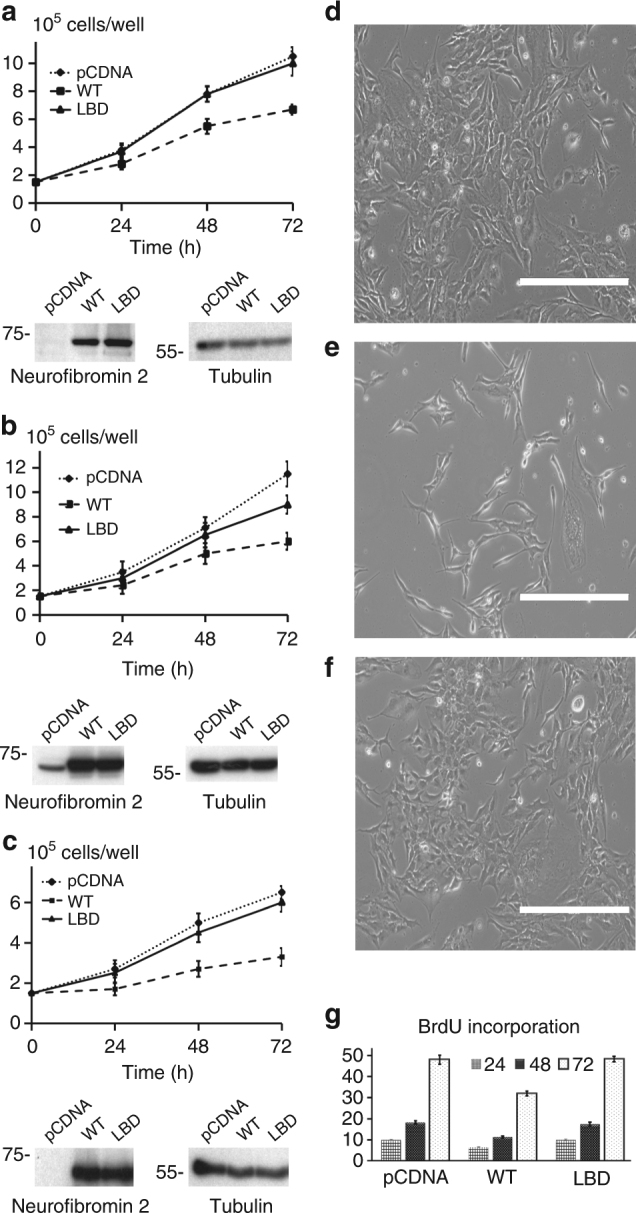
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
