

Mitochondrial Ca2+ Influx Contributes to Arrhythmic Risk in Nonischemic Cardiomyopathy
- PMID: 29627768
- PMCID: PMC6015427
- DOI: 10.1161/JAHA.117.007805
Mitochondrial Ca2+ Influx Contributes to Arrhythmic Risk in Nonischemic Cardiomyopathy
Abstract
Background: Heart failure (HF) is associated with increased arrhythmia risk and triggered activity. Abnormal Ca2+ handling is thought to underlie triggered activity, and mitochondria participate in Ca2+ homeostasis.
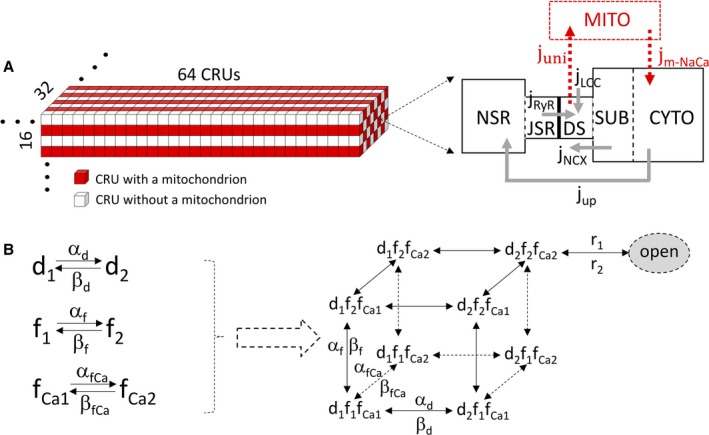
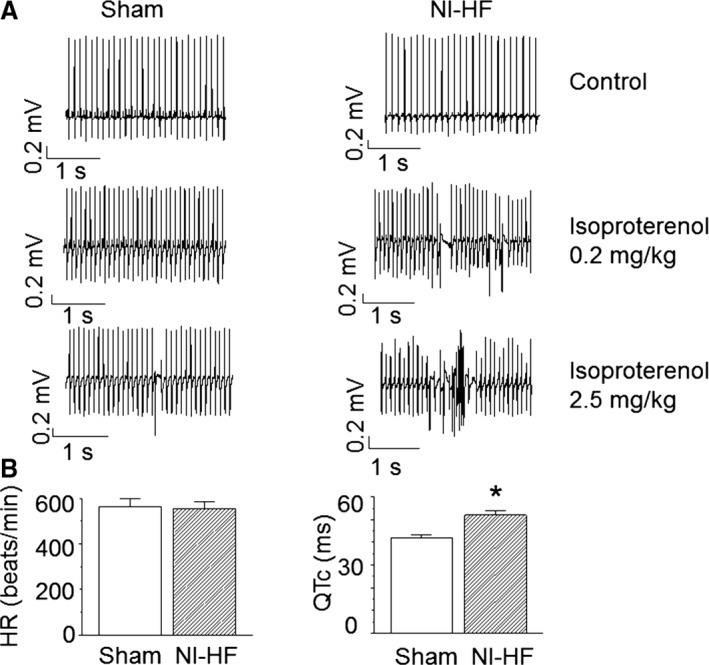
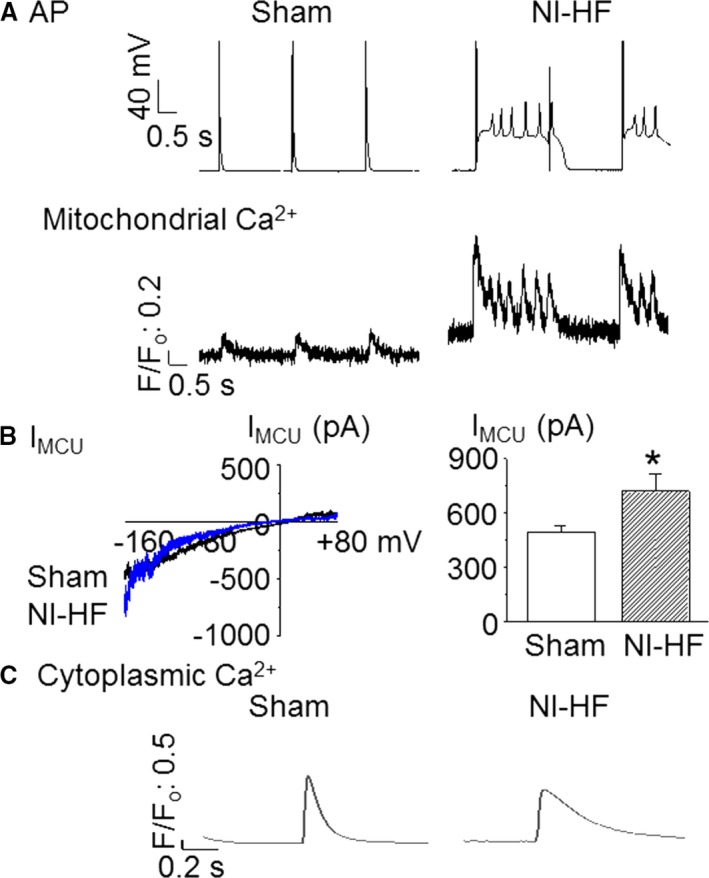
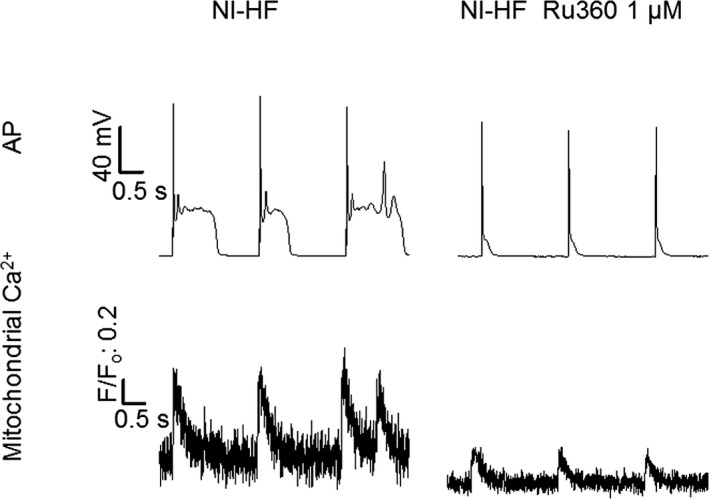
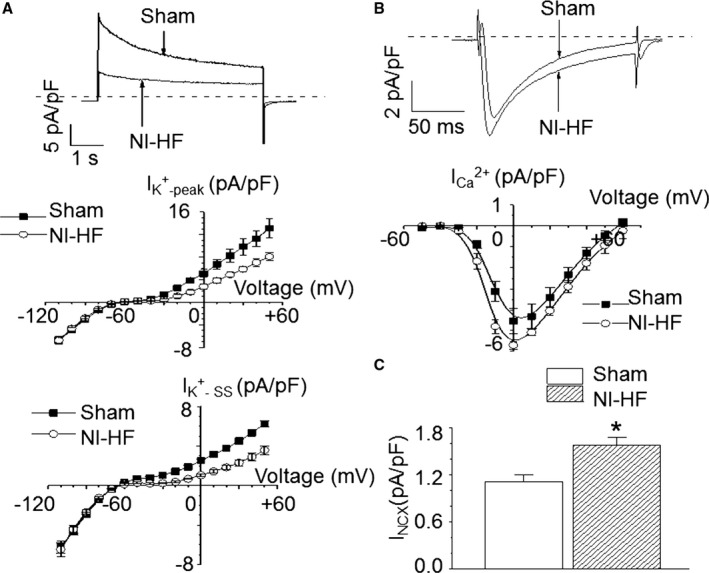
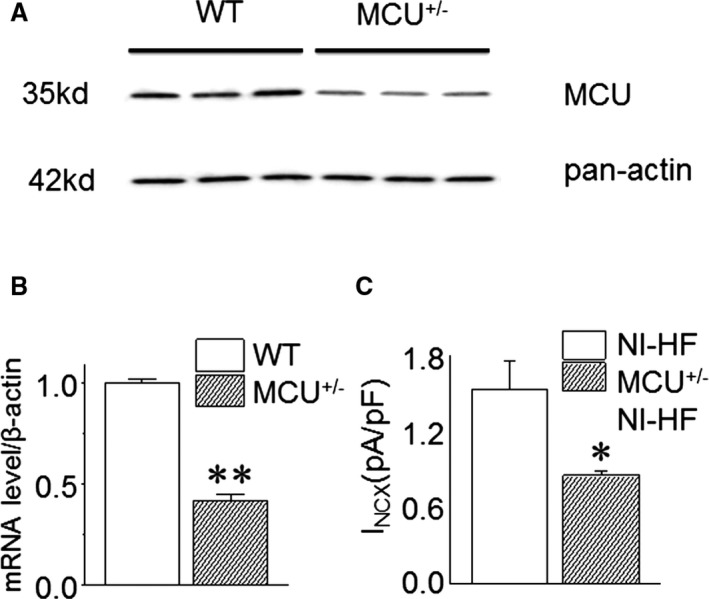
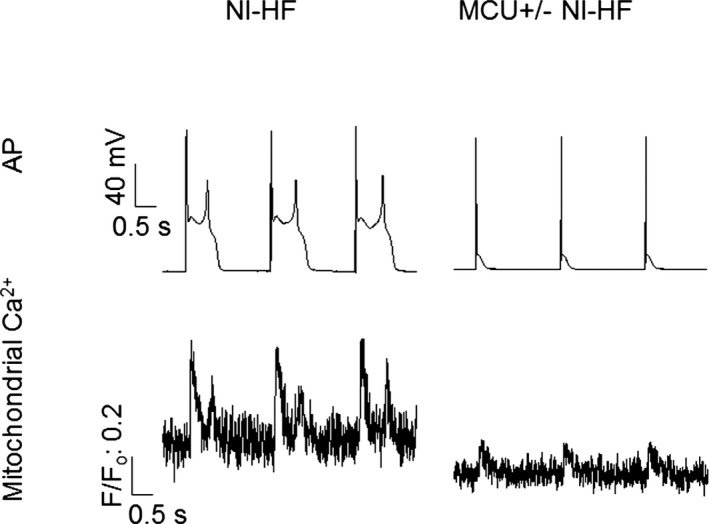
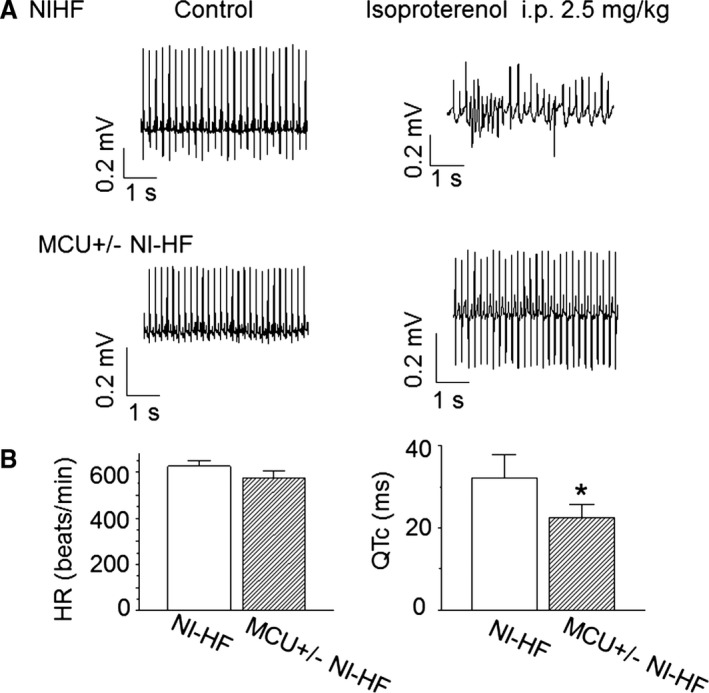
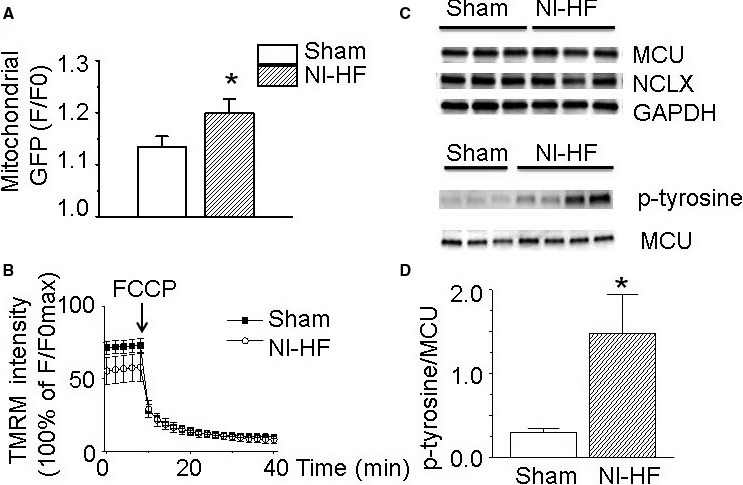
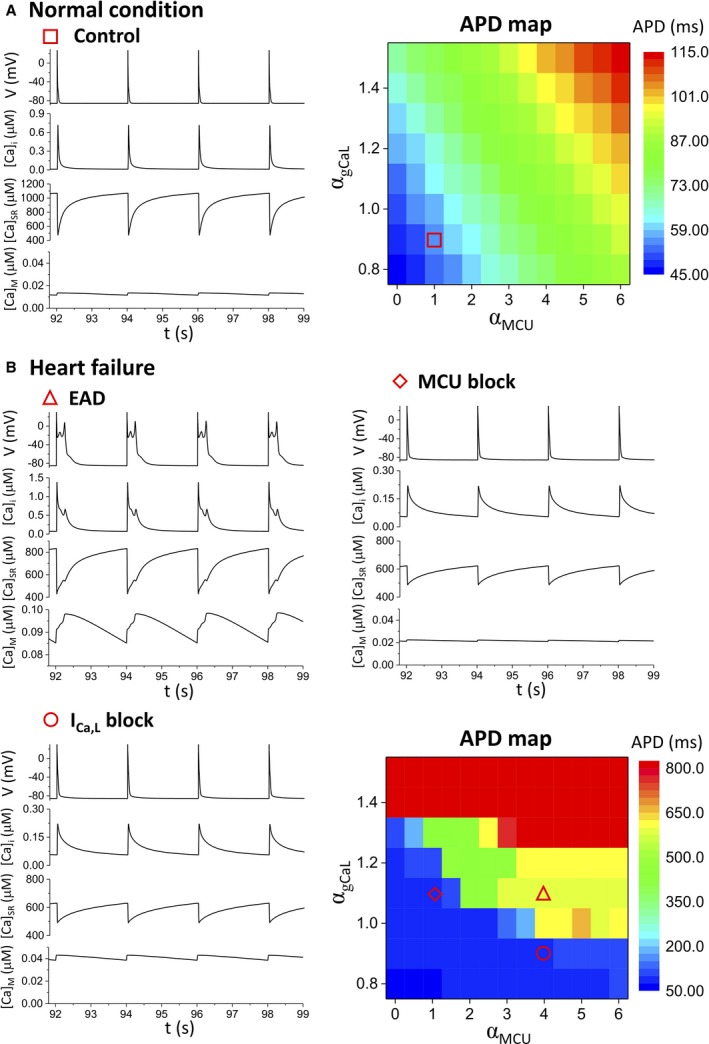
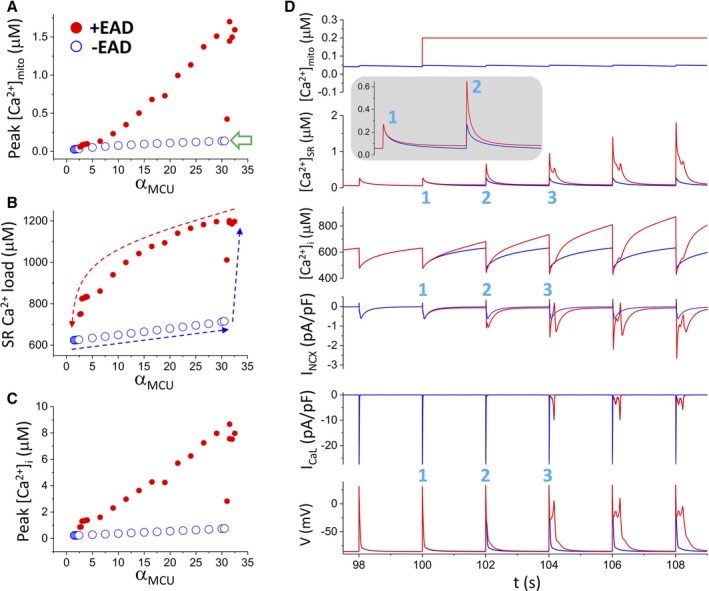
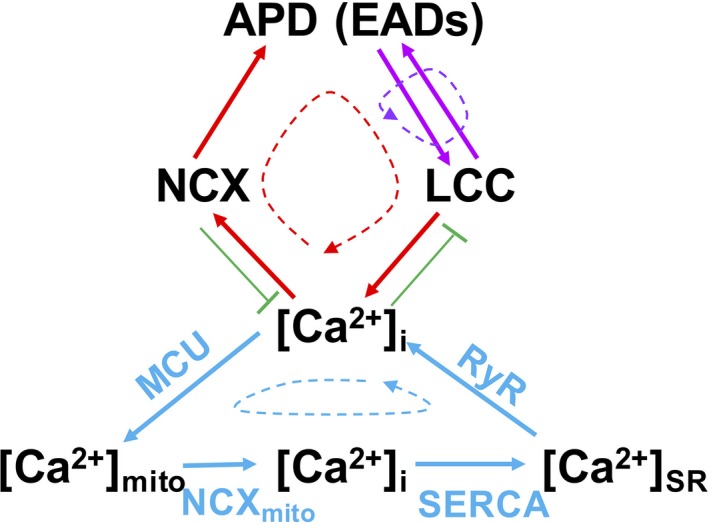
Methods and results: A model of nonischemic HF was induced in C57BL/6 mice by hypertension. Computer simulations were performed using a mouse ventricular myocyte model of HF. Isoproterenol-induced premature ventricular contractions and ventricular fibrillation were more prevalent in nonischemic HF mice than sham controls. Isolated myopathic myocytes showed decreased cytoplasmic Ca2+ transients, increased mitochondrial Ca2+ transients, and increased action potential duration at 90% repolarization. The alteration of action potential duration at 90% repolarization was consistent with in vivo corrected QT prolongation and could be explained by augmented L-type Ca2+ currents, increased Na+-Ca2+ exchange currents, and decreased total K+ currents. Of myopathic ventricular myocytes, 66% showed early afterdepolarizations (EADs) compared with 17% of sham myocytes (P<0.05). Intracellular application of 1 μmol/L Ru360, a mitochondrial Ca2+ uniporter-specific antagonist, could reduce mitochondrial Ca2+ transients, decrease action potential duration at 90% repolarization, and ameliorate EADs. Furthermore, genetic knockdown of mitochondrial Ca2+ uniporters inhibited mitochondrial Ca2+ uptake, reduced Na+-Ca2+ exchange currents, decreased action potential duration at 90% repolarization, suppressed EADs, and reduced ventricular fibrillation in nonischemic HF mice. Computer simulations showed that EADs promoted by HF remodeling could be abolished by blocking either the mitochondrial Ca2+ uniporter or the L-type Ca2+ current, consistent with the experimental observations.
Conclusions: Mitochondrial Ca2+ handling plays an important role in EADs seen with nonischemic cardiomyopathy and may represent a therapeutic target to reduce arrhythmic risk in this condition.
Keywords: arrhythmia; calcium; heart failure; mitochondria.
© 2018 The Authors. Published on behalf of the American Heart Association, Inc., by Wiley.
Figures
References
-
- Nuss HB, Kaab S, Kass DA, Tomaselli GF, Marban E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol. 1999;277:H80–H91. - PubMed
-
- Ruiz‐Meana M, Fernandez‐Sanz C, Garcia‐Dorado D. The SR‐mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc Res. 2010;88:30–39. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
