

Neurodevelopmental Genetic Diseases Associated With Microdeletions and Microduplications of Chromosome 17p13.3
- PMID: 29628935
- PMCID: PMC5876250
- DOI: 10.3389/fgene.2018.00080
Neurodevelopmental Genetic Diseases Associated With Microdeletions and Microduplications of Chromosome 17p13.3
Abstract
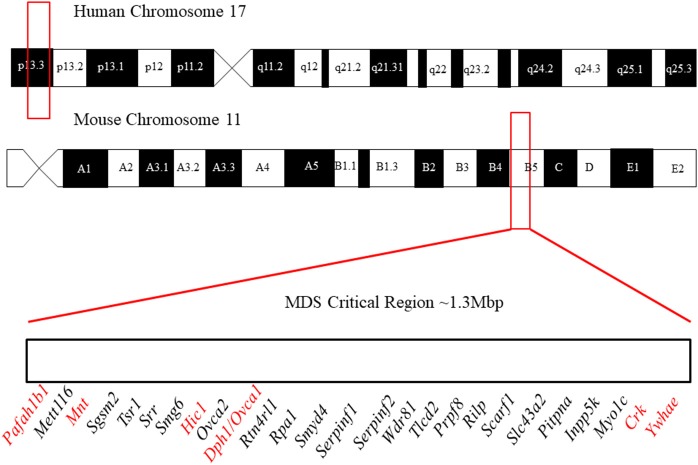
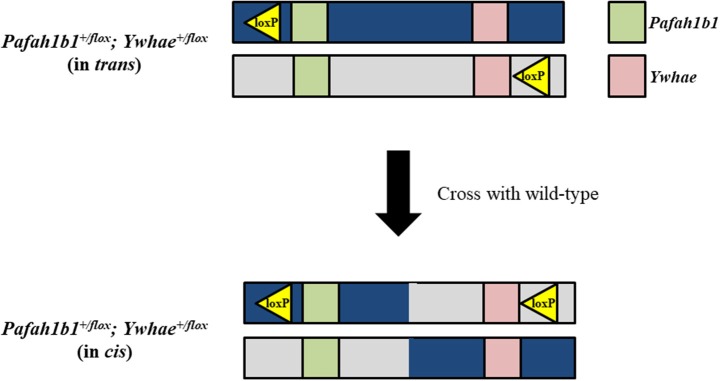
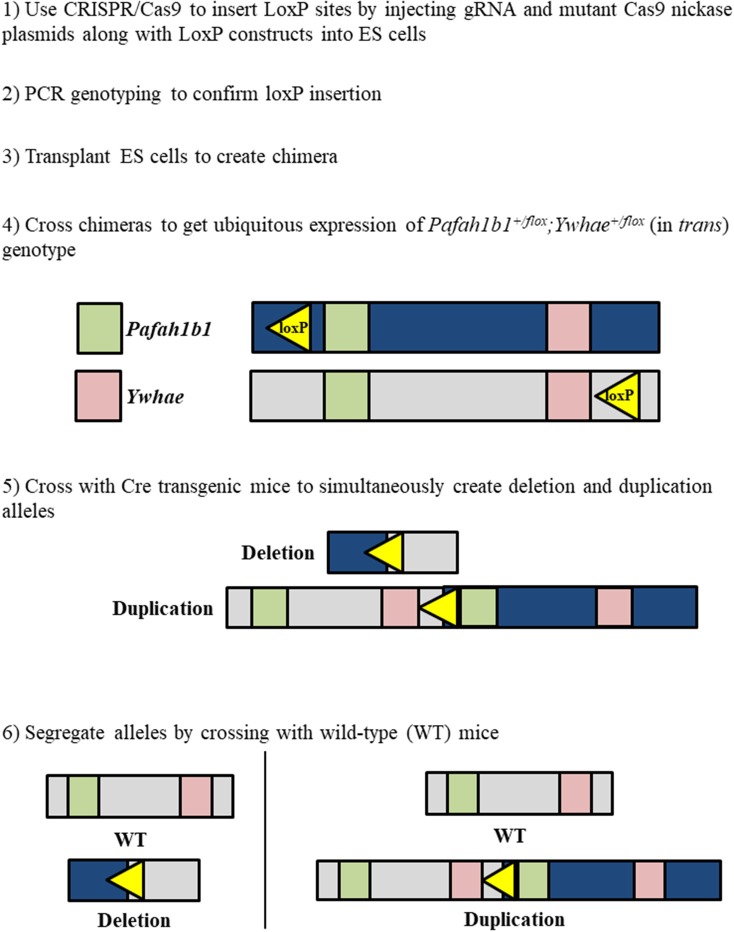
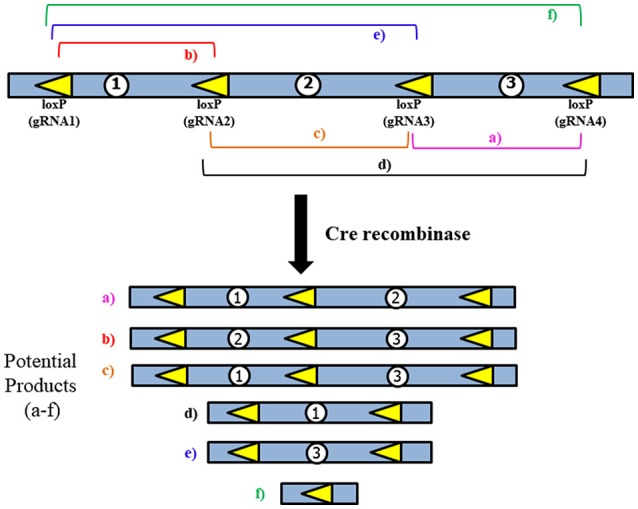
Chromosome 17p13.3 is a region of genomic instability that is linked to different rare neurodevelopmental genetic diseases, depending on whether a deletion or duplication of the region has occurred. Chromosome microdeletions within 17p13.3 can result in either isolated lissencephaly sequence (ILS) or Miller-Dieker syndrome (MDS). Both conditions are associated with a smooth cerebral cortex, or lissencephaly, which leads to developmental delay, intellectual disability, and seizures. However, patients with MDS have larger deletions than patients with ILS, resulting in additional symptoms such as poor muscle tone, congenital anomalies, abnormal spasticity, and craniofacial dysmorphisms. In contrast to microdeletions in 17p13.3, recent studies have attracted considerable attention to a condition known as a 17p13.3 microduplication syndrome. Depending on the genes involved in their microduplication, patients with 17p13.3 microduplication syndrome may be categorized into either class I or class II. Individuals in class I have microduplications of the YWHAE gene encoding 14-3-3ε, as well as other genes in the region. However, the PAFAH1B1 gene encoding LIS1 is never duplicated in these patients. Class I microduplications generally result in learning disabilities, autism, and developmental delays, among other disorders. Individuals in class II always have microduplications of the PAFAH1B1 gene, which may include YWHAE and other genetic microduplications. Class II microduplications generally result in smaller body size, developmental delays, microcephaly, and other brain malformations. Here, we review the phenotypes associated with copy number variations (CNVs) of chromosome 17p13.3 and detail their developmental connection to particular microdeletions or microduplications. We also focus on existing single and double knockout mouse models that have been used to study human phenotypes, since the highly limited number of patients makes a study of these conditions difficult in humans. These models are also crucial for the study of brain development at a mechanistic level since this cannot be accomplished in humans. Finally, we emphasize the usefulness of the CRISPR/Cas9 system and next generation sequencing in the study of neurodevelopmental diseases.
Keywords: 17p13.3; CRISPR; autism spectrum disorder; lissencephaly; microdeletion; microduplication; neurodevelopmental disorder; next generation sequence.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous