

Selective Genetic Overlap Between Amyotrophic Lateral Sclerosis and Diseases of the Frontotemporal Dementia Spectrum
- PMID: 29630712
- PMCID: PMC6043387
- DOI: 10.1001/jamaneurol.2018.0372
Selective Genetic Overlap Between Amyotrophic Lateral Sclerosis and Diseases of the Frontotemporal Dementia Spectrum
Abstract
Importance: Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder characterized by loss of upper and lower motor neurons. Although novel ALS genetic variants have been identified, the shared genetic risk between ALS and other neurodegenerative disorders remains poorly understood.
Objectives: To examine whether there are common genetic variants that determine the risk for ALS and other neurodegenerative diseases and to identify their functional pathways.
Design, setting, and participants: In this study conducted from December 1, 2016, to August 1, 2017, the genetic overlap between ALS, sporadic frontotemporal dementia (FTD), FTD with TDP-43 inclusions, Parkinson disease (PD), Alzheimer disease (AD), corticobasal degeneration (CBD), and progressive supranuclear palsy (PSP) were systematically investigated in 124 876 cases and controls. No participants were excluded from this study. Diagnoses were established using consensus criteria.
Main outcomes and measures: The primary outcomes were a list of novel loci and their functional pathways in ALS, FTD, PSP, and ALS mouse models.
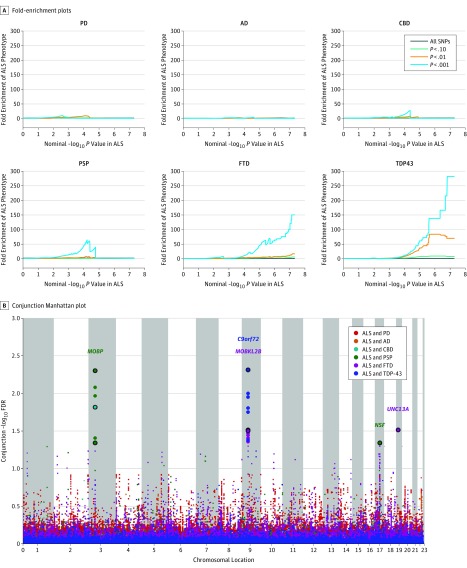
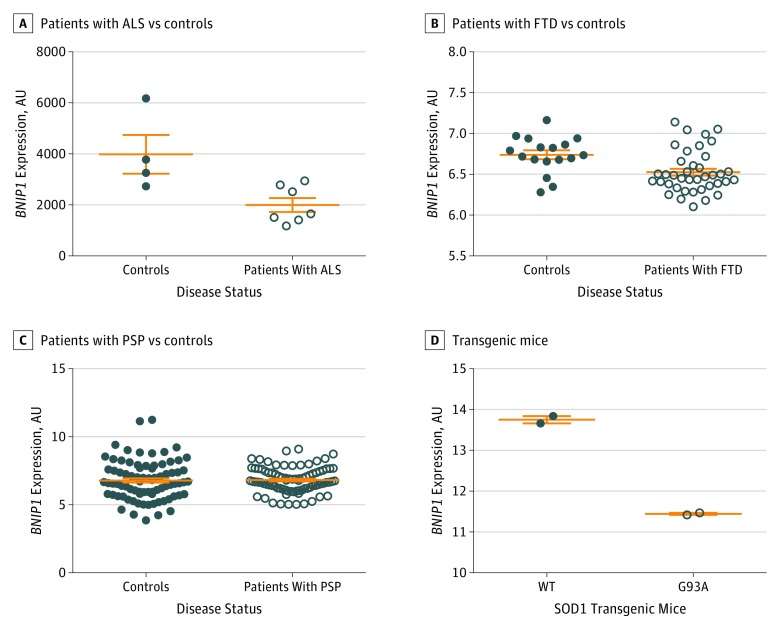
Results: Among 124 876 cases and controls, genome-wide conjunction analyses of ALS, FTD, PD, AD, CBD, and PSP revealed significant genetic overlap between ALS and FTD at known ALS loci: rs13302855 and rs3849942 (nearest gene, C9orf72; P = .03 for rs13302855 and P = .005 for rs3849942) and rs4239633 (nearest gene, UNC13A; P = .03). Significant genetic overlap was also found between ALS and PSP at rs7224296, which tags the MAPT H1 haplotype (nearest gene, NSF; P = .045). Shared risk genes were enriched for pathways involving neuronal function and development. At a conditional FDR P < .05, 22 novel ALS polymorphisms were found, including rs538622 (nearest gene, ERGIC1; P = .03 for ALS and FTD), which modifies BNIP1 expression in human brains (35 of 137 females; mean age, 59 years; P = .001). BNIP1 expression was significantly reduced in spinal cord motor neurons from patients with ALS (4 controls: mean age, 60.5 years, mean [SE] value, 3984 [760.8] arbitrary units [AU]; 7 patients with ALS: mean age, 56 years, mean [SE] value, 1999 [274.1] AU; P = .02), in an ALS mouse model (mean [SE] value, 13.75 [0.09] AU for 2 SOD1 WT mice and 11.45 [0.03] AU for 2 SOD1 G93A mice; P = .002) and in brains of patients with PSP (80 controls: 39 females; mean age, 82 years, mean [SE] value, 6.8 [0.2] AU; 84 patients with PSP: 33 females, mean age 74 years, mean [SE] value, 6.8 [0.1] AU; β = -0.19; P = .009) or FTD (11 controls: 4 females; mean age, 67 years; mean [SE] value, 6.74 [0.05] AU; 17 patients with FTD: 10 females; mean age, 69 years; mean [SE] value, 6.53 [0.04] AU; P = .005).
Conclusions and relevance: This study found novel genetic overlap between ALS and diseases of the FTD spectrum, that the MAPT H1 haplotype confers risk for ALS, and identified the mitophagy-associated, proapoptotic protein BNIP1 as an ALS risk gene. Together, these findings suggest that sporadic ALS may represent a selectively pleiotropic, polygenic disorder.
Conflict of interest statement
Figures
Similar articles
-
Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia.Acta Neuropathol. 2017 May;133(5):825-837. doi: 10.1007/s00401-017-1693-y. Epub 2017 Mar 7. Acta Neuropathol. 2017. PMID: 28271184 Free PMC article.
-
C9orf72 repeat expansions are restricted to the ALS-FTD spectrum.Neurobiol Aging. 2014 Apr;35(4):936.e13-7. doi: 10.1016/j.neurobiolaging.2013.09.037. Epub 2013 Oct 2. Neurobiol Aging. 2014. PMID: 24169076
-
C9orf72 and UNC13A are shared risk loci for amyotrophic lateral sclerosis and frontotemporal dementia: a genome-wide meta-analysis.Ann Neurol. 2014 Jul;76(1):120-33. doi: 10.1002/ana.24198. Epub 2014 Jun 27. Ann Neurol. 2014. PMID: 24931836 Free PMC article.
-
[Genetic architecture of amyotrophic lateral sclerosis and frontotemporal dementia : Overlap and differences].Nervenarzt. 2017 Jul;88(7):728-735. doi: 10.1007/s00115-017-0349-4. Nervenarzt. 2017. PMID: 28573364 Review. German.
-
Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD.Brain Res. 2018 Aug 15;1693(Pt A):98-108. doi: 10.1016/j.brainres.2018.02.011. Epub 2018 Feb 14. Brain Res. 2018. PMID: 29453960 Free PMC article. Review.
Cited by
-
Neuroimaging in genetic frontotemporal dementia and amyotrophic lateral sclerosis.Neurobiol Dis. 2020 Nov;145:105063. doi: 10.1016/j.nbd.2020.105063. Epub 2020 Sep 2. Neurobiol Dis. 2020. PMID: 32890771 Free PMC article. Review.
-
Shared genetic architecture between leukocyte telomere length and Alzheimer's disease.Alzheimers Res Ther. 2025 May 17;17(1):108. doi: 10.1186/s13195-025-01757-z. Alzheimers Res Ther. 2025. PMID: 40382655 Free PMC article.
-
Axonal Endoplasmic Reticulum Dynamics and Its Roles in Neurodegeneration.Front Neurosci. 2020 Jan 29;14:48. doi: 10.3389/fnins.2020.00048. eCollection 2020. Front Neurosci. 2020. PMID: 32116502 Free PMC article. Review.
-
Accurate error control in high-dimensional association testing using conditional false discovery rates.Biom J. 2021 Jun;63(5):1096-1130. doi: 10.1002/bimj.201900254. Epub 2021 Mar 7. Biom J. 2021. PMID: 33682201 Free PMC article.
-
Non-coding variation in dementias: mechanisms, insights, and challenges.NPJ Dement. 2025;1(1):9. doi: 10.1038/s44400-025-00012-4. Epub 2025 Jun 3. NPJ Dement. 2025. PMID: 40476256 Free PMC article. Review.
References
-
- Cairns NJ, Bigio EH, Mackenzie IRA, et al. ; Consortium for Frontotemporal Lobar Degeneration . Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114(1):5-22. - PMC - PubMed
-
- Neumann M, Sampathu DM, Kwong LK, et al. . Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130-133. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
