

Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis
- PMID: 29632024
- PMCID: PMC6034641
- DOI: 10.1182/blood-2017-11-814244
Genetic and mechanistic diversity in pediatric hemophagocytic lymphohistiocytosis
Abstract
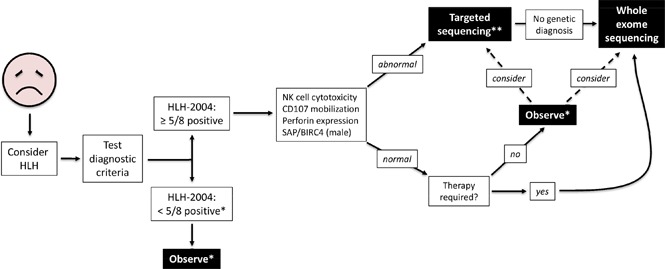
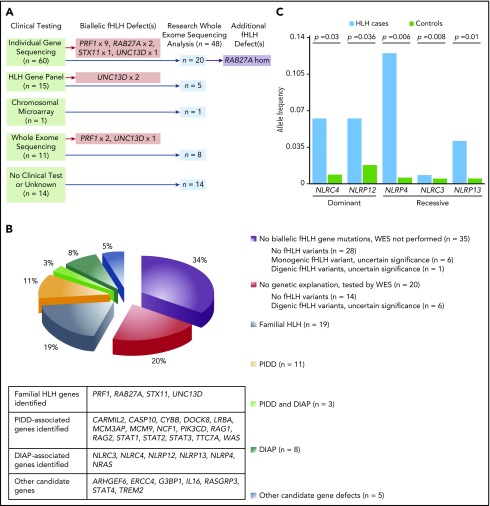
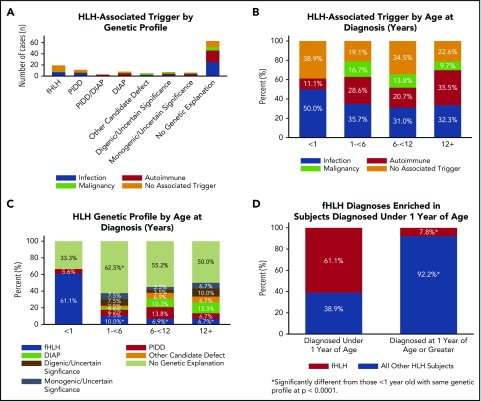
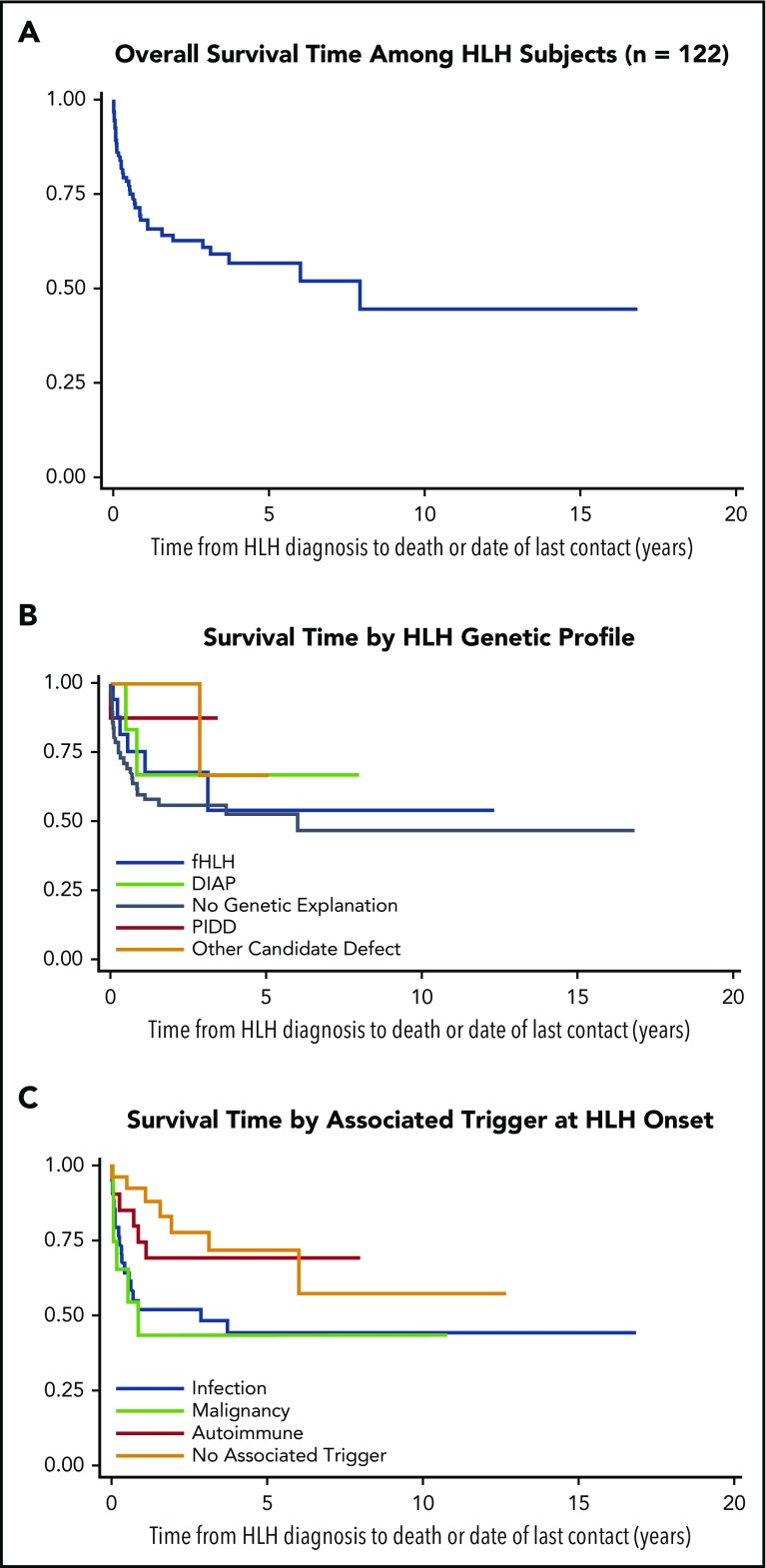
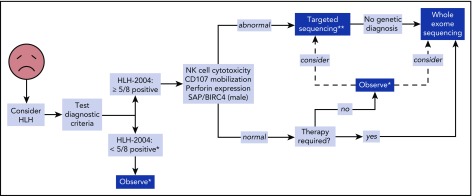
The HLH-2004 criteria are used to diagnose hemophagocytic lymphohistiocytosis (HLH), yet concern exists for their misapplication, resulting in suboptimal treatment of some patients. We sought to define the genomic spectrum and associated outcomes of a diverse cohort of children who met the HLH-2004 criteria. Genetic testing was performed clinically or through research-based whole-exome sequencing. Clinical metrics were analyzed with respect to genomic results. Of 122 subjects enrolled over the course of 17 years, 101 subjects received genetic testing. Biallelic familial HLH (fHLH) gene defects were identified in only 19 (19%) and correlated with presentation at younger than 1 year of age (P < .0001). Digenic fHLH variants were observed but lacked statistical support for disease association. In 28 (58%) of 48 subjects, research whole-exome sequencing analyses successfully identified likely molecular explanations, including underlying primary immunodeficiency diseases, dysregulated immune activation and proliferation disorders, and potentially novel genetic conditions. Two-thirds of patients identified by the HLH-2004 criteria had underlying etiologies for HLH, including genetic defects, autoimmunity, and malignancy. Overall survival was 45%, and increased mortality correlated with HLH triggered by infection or malignancy (P < .05). Differences in survival did not correlate with genetic profile or extent of therapy. HLH should be conceptualized as a phenotype of critical illness characterized by toxic activation of immune cells from different underlying mechanisms. In most patients with HLH, targeted sequencing of fHLH genes remains insufficient for identifying pathogenic mechanisms. Whole-exome sequencing, however, may identify specific therapeutic opportunities and affect hematopoietic stem cell transplantation options for these patients.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
Comment in
-
HLH: genomics illuminates pathophysiological diversity.Blood. 2018 Jul 5;132(1):5-7. doi: 10.1182/blood-2018-05-845818. Blood. 2018. PMID: 29976779 No abstract available.
References
-
- Bodley Scott R, Robb-Smith AHT. Histiocytic medullary reticulosis. Lancet. 1939;234(6047):194-198.
-
- Henter J-I, Horne A, Aricó M, et al. . HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124-131. - PubMed
-
- Allen CE, McClain KL. Pathophysiology and epidemiology of hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ Program. 2015;2015:177-182. - PubMed
-
- Henter J-I, Samuelsson-Horne A, Aricò M, et al. ; Histocyte Society. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-2373. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
