

SNX14 mutations affect endoplasmic reticulum-associated neutral lipid metabolism in autosomal recessive spinocerebellar ataxia 20
- PMID: 29635513
- PMCID: PMC5961352
- DOI: 10.1093/hmg/ddy101
SNX14 mutations affect endoplasmic reticulum-associated neutral lipid metabolism in autosomal recessive spinocerebellar ataxia 20
Abstract
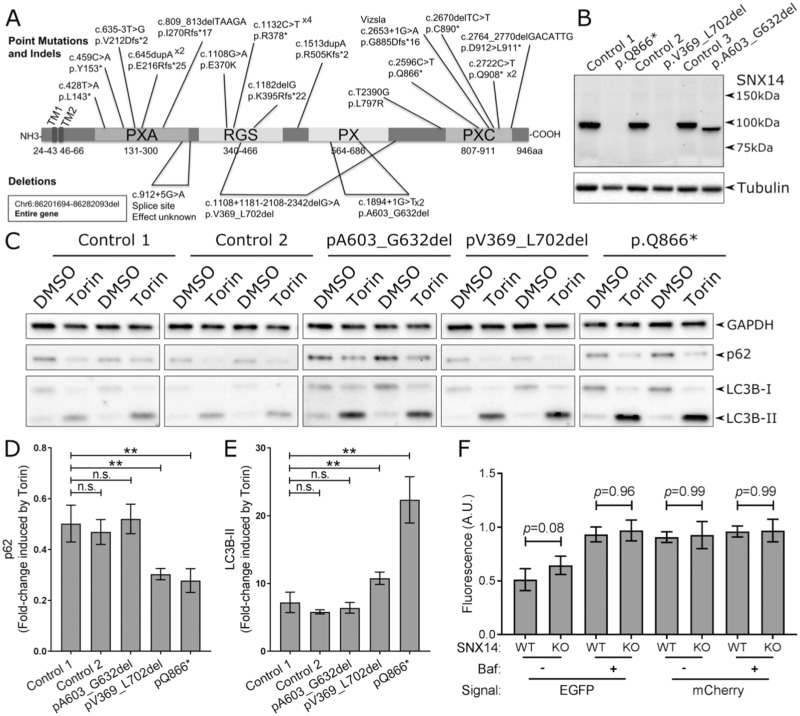
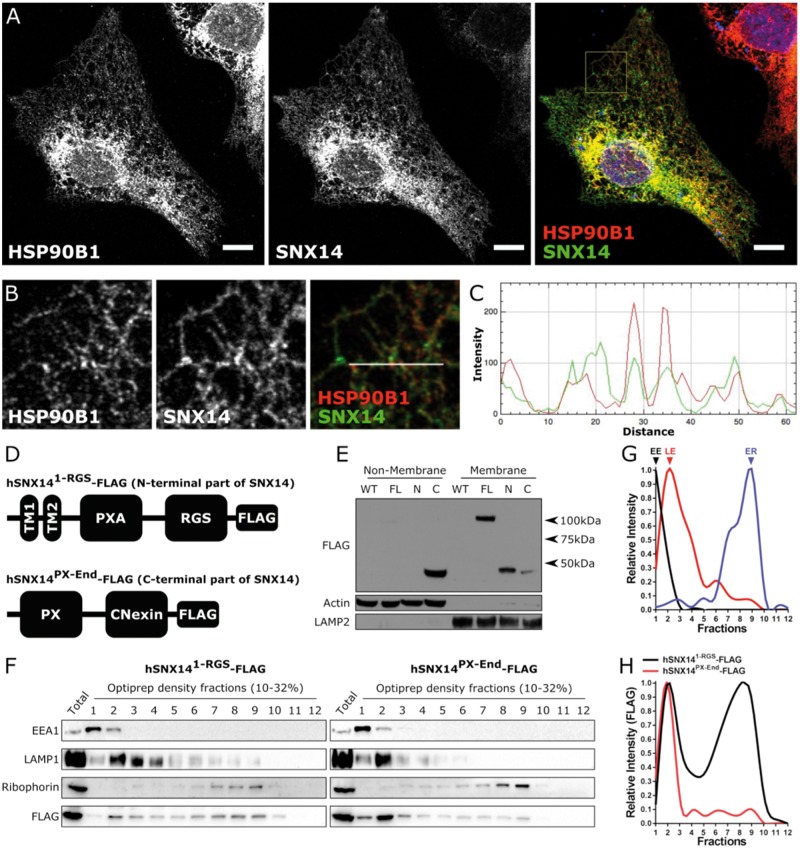
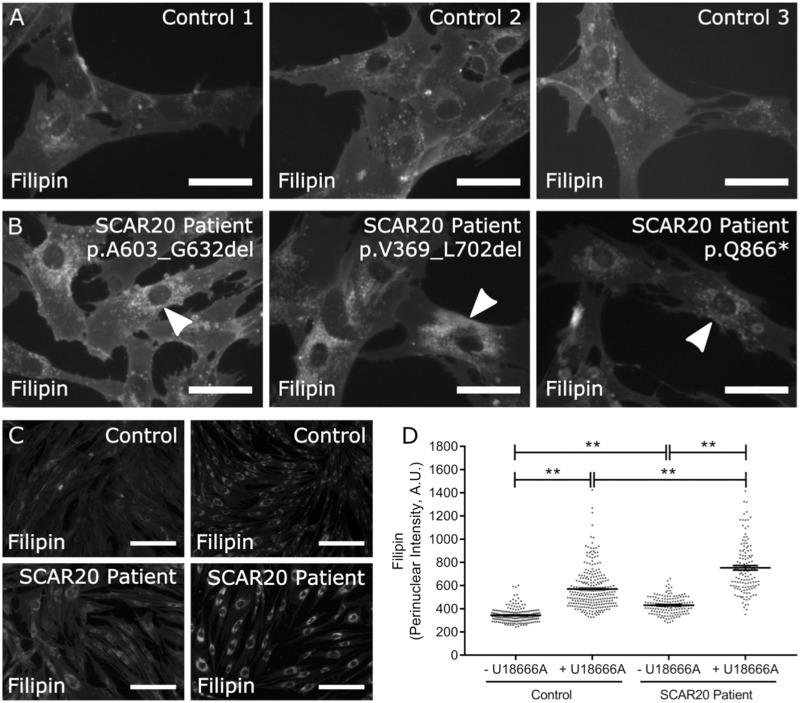
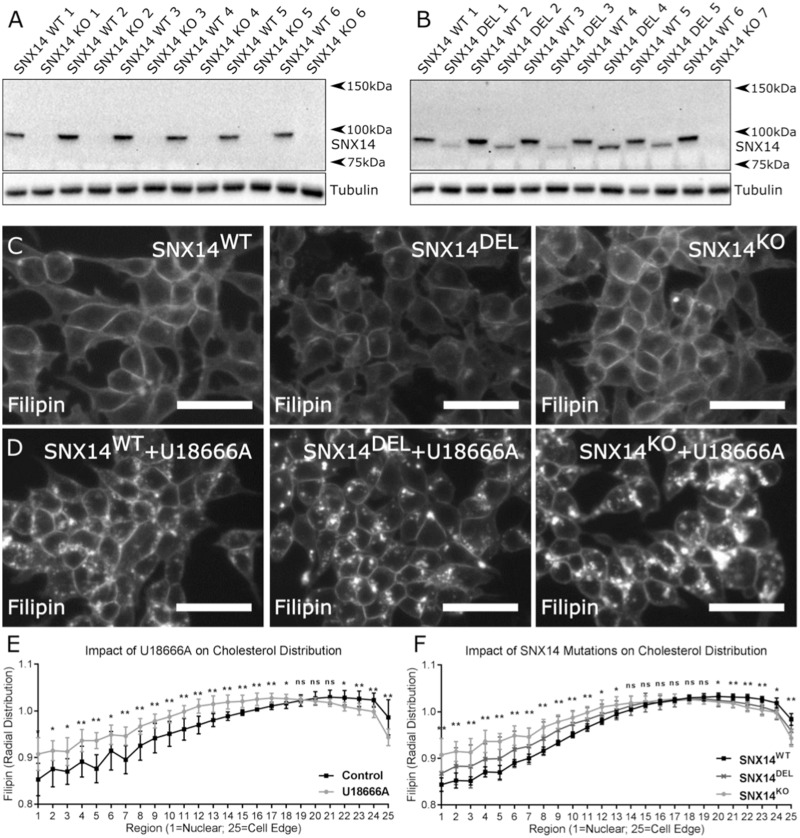
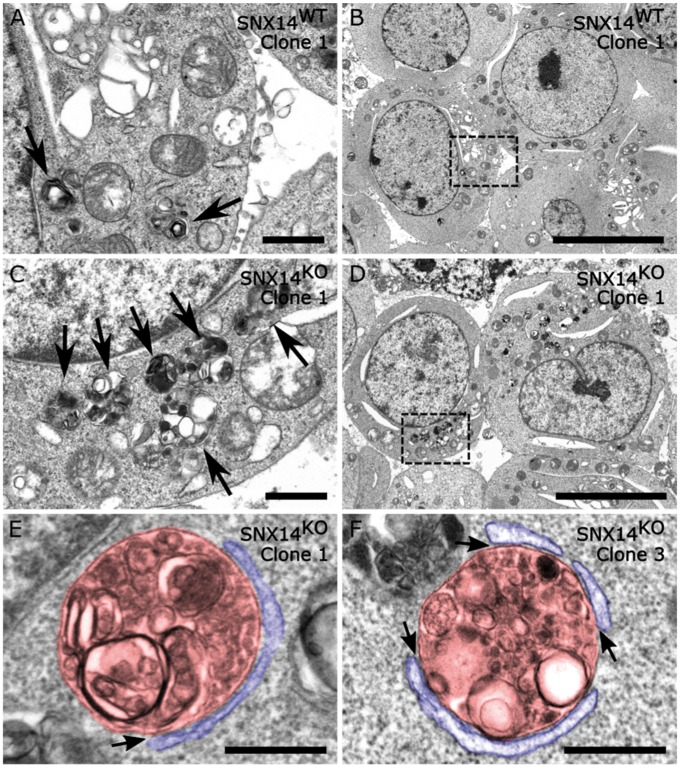
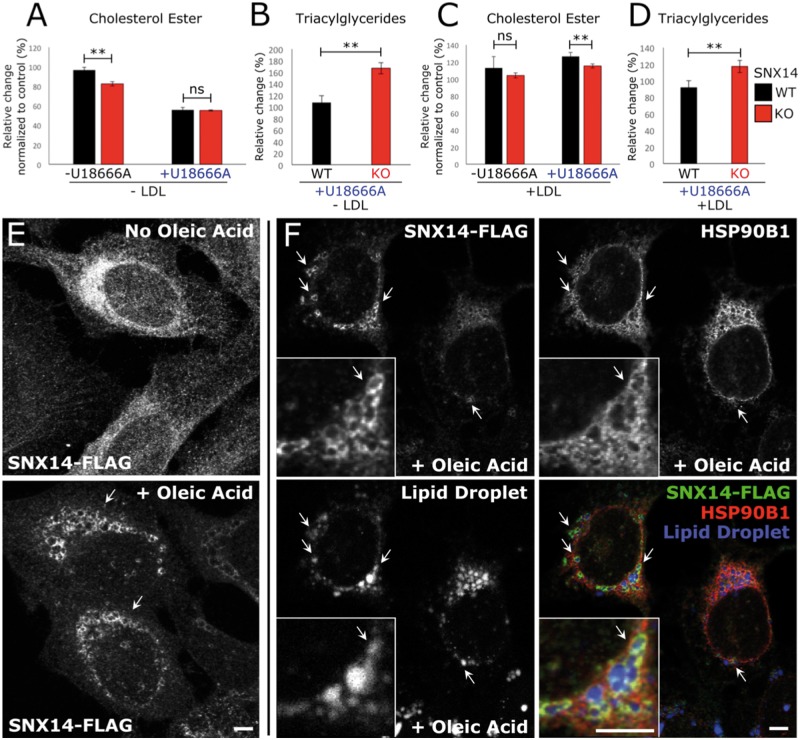
Mutations in SNX14 cause the autosomal recessive cerebellar ataxia 20 (SCAR20). Mutations generally result in loss of protein although several coding region deletions have also been reported. Patient-derived fibroblasts show disrupted autophagy, but the precise function of SNX14 is unknown. The yeast homolog, Mdm1, functions in endoplasmic reticulum (ER)-lysosome/vacuole inter-organelle tethering, but functional conservation in mammals is still required. Here, we show that loss of SNX14 alters but does not block autophagic flux. In addition, we find that SNX14 is an ER-associated protein that functions in neutral lipid homeostasis and inter-organelle crosstalk. SNX14 requires its N-terminal transmembrane helices for ER localization, while the Phox homology (PX) domain is dispensable for subcellular localization. Both SNX14-mutant fibroblasts and SNX14KO HEK293 cells accumulate aberrant cytoplasmic vacuoles, suggesting defects in endolysosomal homeostasis. However, ER-late endosome/lysosome contact sites are maintained in SNX14KO cells, indicating that it is not a prerequisite for ER-endolysosomal tethering. Further investigation of SNX14- deficiency indicates general defects in neutral lipid metabolism. SNX14KO cells display distinct perinuclear accumulation of filipin in LAMP1-positive lysosomal structures indicating cholesterol accumulation. Consistent with this, SNX14KO cells display a slight but detectable decrease in cholesterol ester levels, which is exacerbated with U18666A. Finally, SNX14 associates with ER-derived lipid droplets (LD) following oleate treatment, indicating a role in ER-LD crosstalk. We therefore identify an important role for SNX14 in neutral lipid homeostasis between the ER, lysosomes and LDs that may provide an early intervention target to alleviate the clinical symptoms of SCAR20.
Figures
References
-
- Thomas A.C., Williams H., Seto-Salvia N., Bacchelli C., Jenkins D., O'Sullivan M., Mengrelis K., Ishida M., Ocaka L., Chanudet E.. et al. (2014) Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am. J. Hum. Genet., 95, 611–621. - PMC - PubMed
-
- Jazayeri R., Hu H., Fattahi Z., Musante L., Abedini S.S., Hosseini M., Wienker T.F., Ropers H.H., Najmabadi H., Kahrizi K. (2015) Exome sequencing and linkage analysis identified novel candidate genes in recessive intellectual disability associated with ataxia. Arch. Iranian Med., 18, 670–682. - PubMed
-
- Karaca E., Harel T., Pehlivan D., Jhangiani S.N., Gambin T., Coban Akdemir Z., Gonzaga-Jauregui C., Erdin S., Bayram Y., Campbell I.M.. et al. (2015) Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron, 88, 499–513. - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
