

Regulation of Tumor Progression by Programmed Necrosis
- PMID: 29636841
- PMCID: PMC5831895
- DOI: 10.1155/2018/3537471
Regulation of Tumor Progression by Programmed Necrosis
Abstract
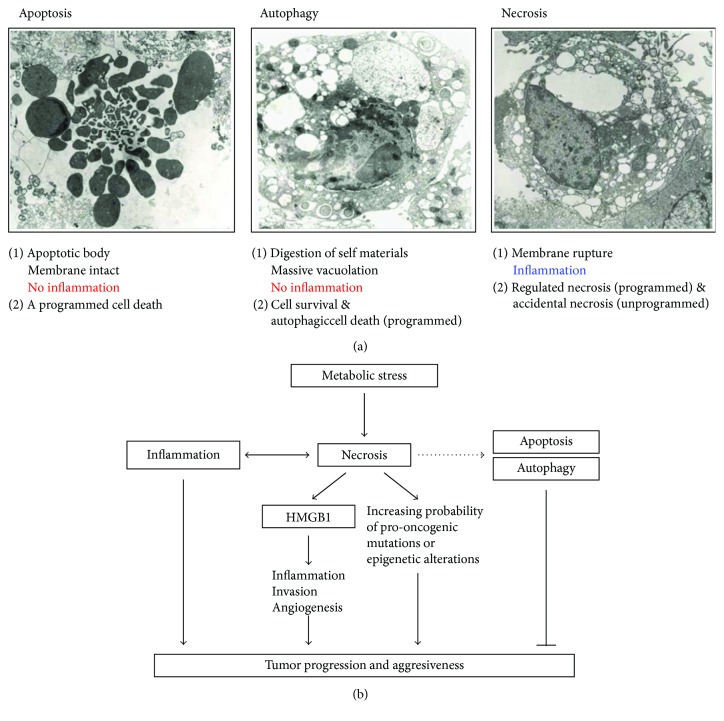
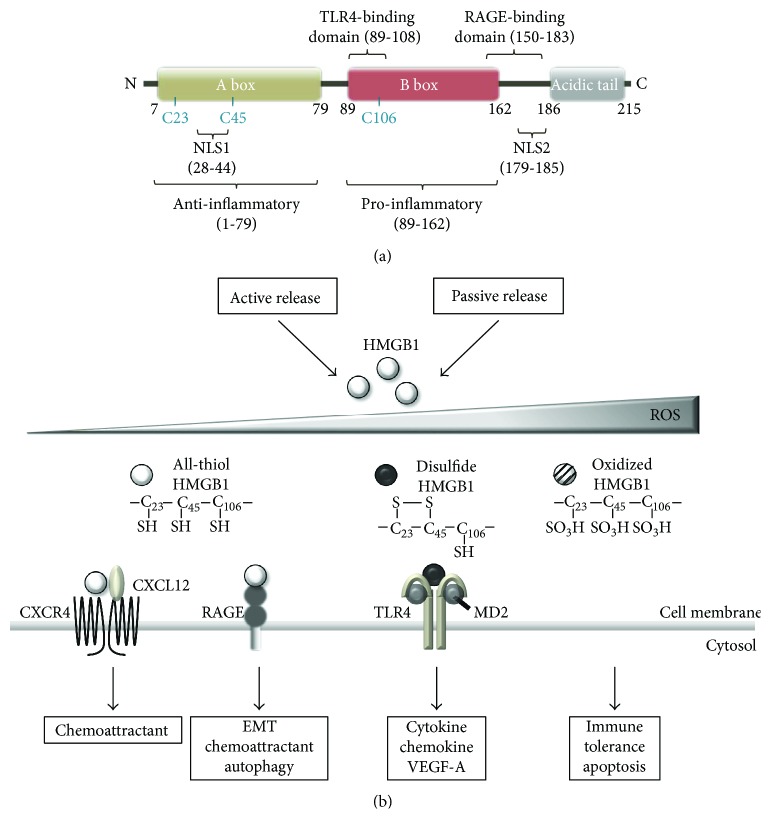
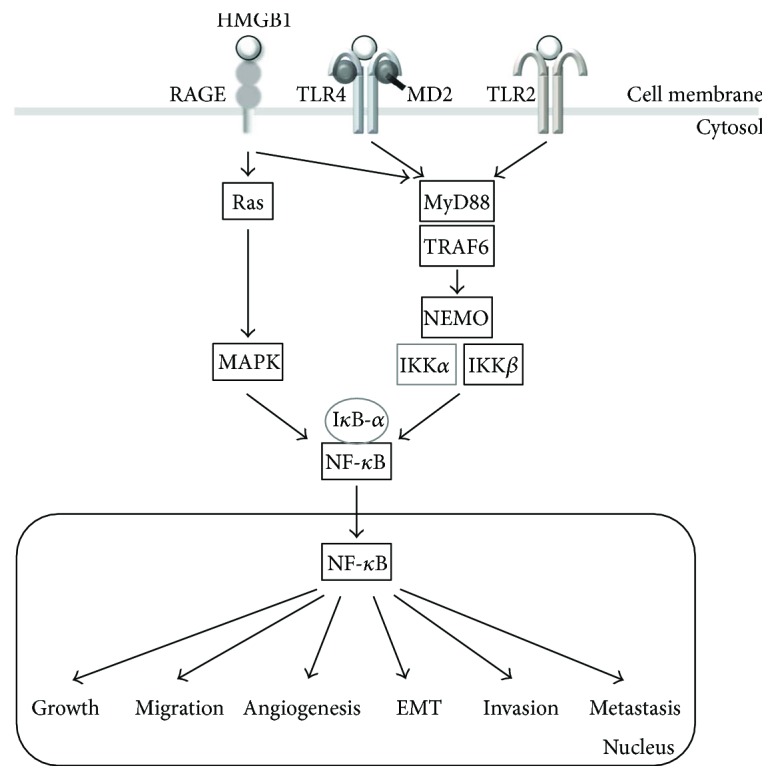
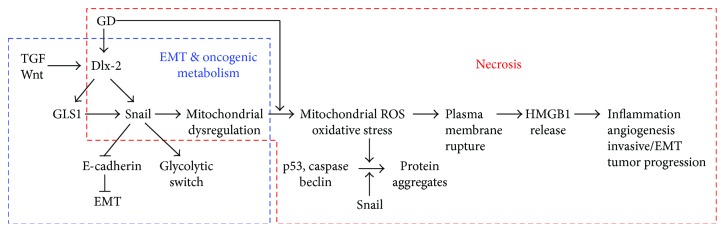
Rapidly growing malignant tumors frequently encounter hypoxia and nutrient (e.g., glucose) deprivation, which occurs because of insufficient blood supply. This results in necrotic cell death in the core region of solid tumors. Necrotic cells release their cellular cytoplasmic contents into the extracellular space, such as high mobility group box 1 (HMGB1), which is a nonhistone nuclear protein, but acts as a proinflammatory and tumor-promoting cytokine when released by necrotic cells. These released molecules recruit immune and inflammatory cells, which exert tumor-promoting activity by inducing angiogenesis, proliferation, and invasion. Development of a necrotic core in cancer patients is also associated with poor prognosis. Conventionally, necrosis has been thought of as an unregulated process, unlike programmed cell death processes like apoptosis and autophagy. Recently, necrosis has been recognized as a programmed cell death, encompassing processes such as oncosis, necroptosis, and others. Metabolic stress-induced necrosis and its regulatory mechanisms have been poorly investigated until recently. Snail and Dlx-2, EMT-inducing transcription factors, are responsible for metabolic stress-induced necrosis in tumors. Snail and Dlx-2 contribute to tumor progression by promoting necrosis and inducing EMT and oncogenic metabolism. Oncogenic metabolism has been shown to play a role(s) in initiating necrosis. Here, we discuss the molecular mechanisms underlying metabolic stress-induced programmed necrosis that promote tumor progression and aggressiveness.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
