

An efficient and scalable pipeline for epitope tagging in mammalian stem cells using Cas9 ribonucleoprotein
- PMID: 29638216
- PMCID: PMC5947990
- DOI: 10.7554/eLife.35069
An efficient and scalable pipeline for epitope tagging in mammalian stem cells using Cas9 ribonucleoprotein
Abstract
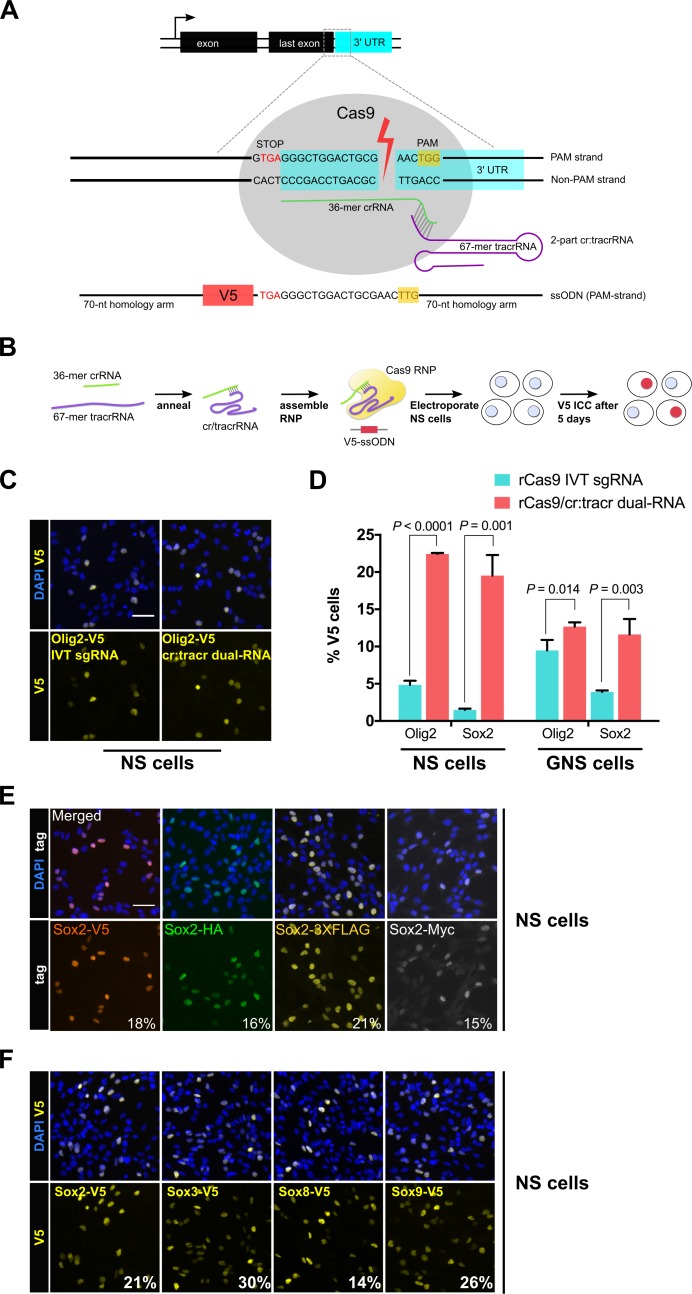
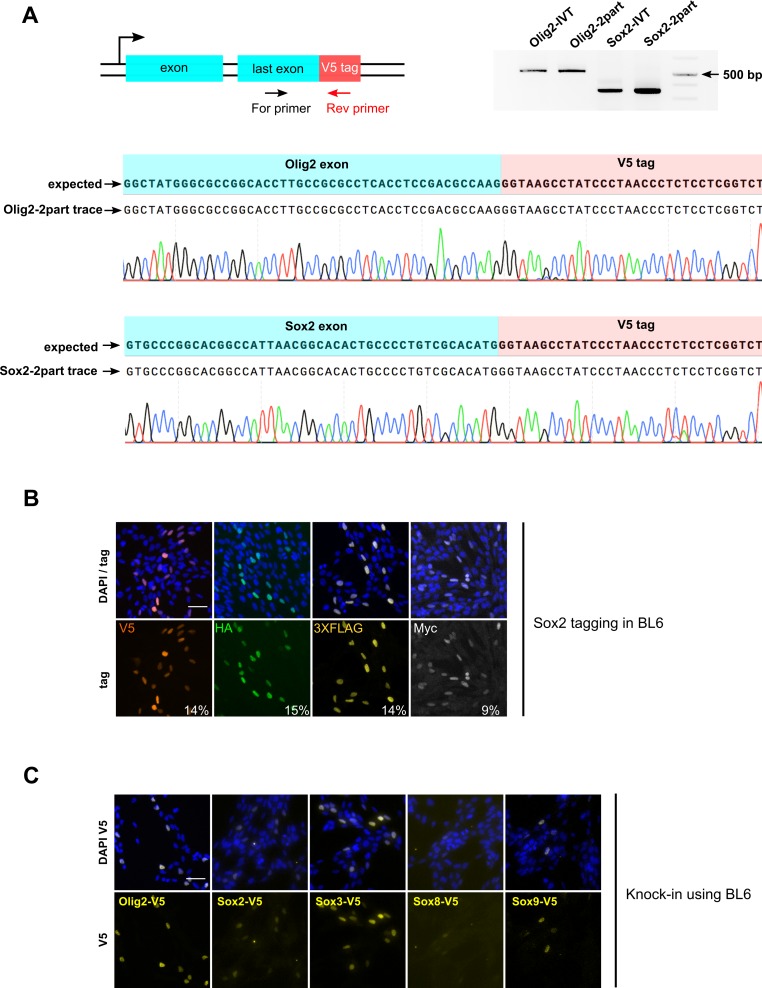
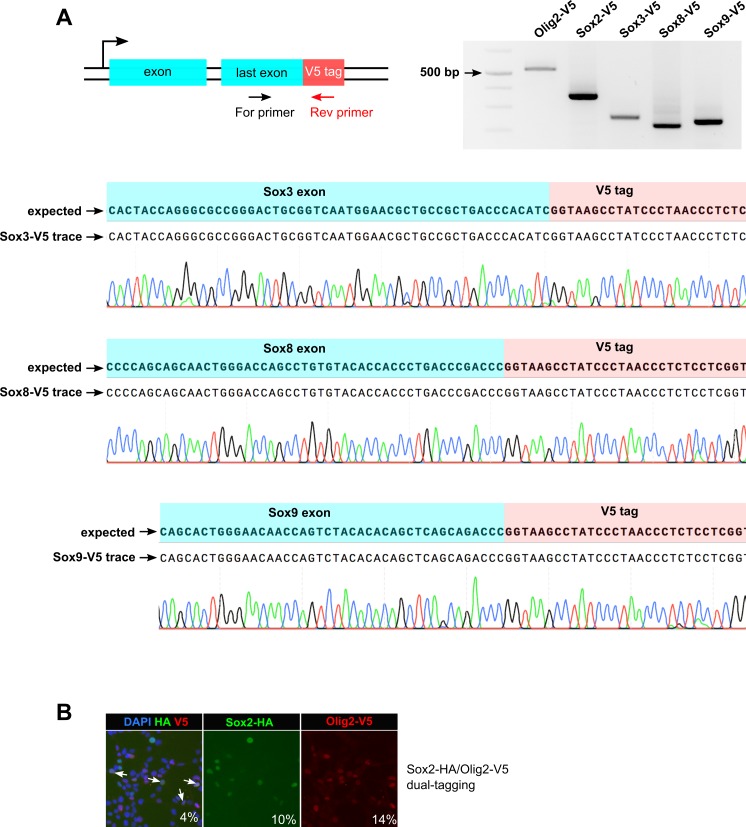
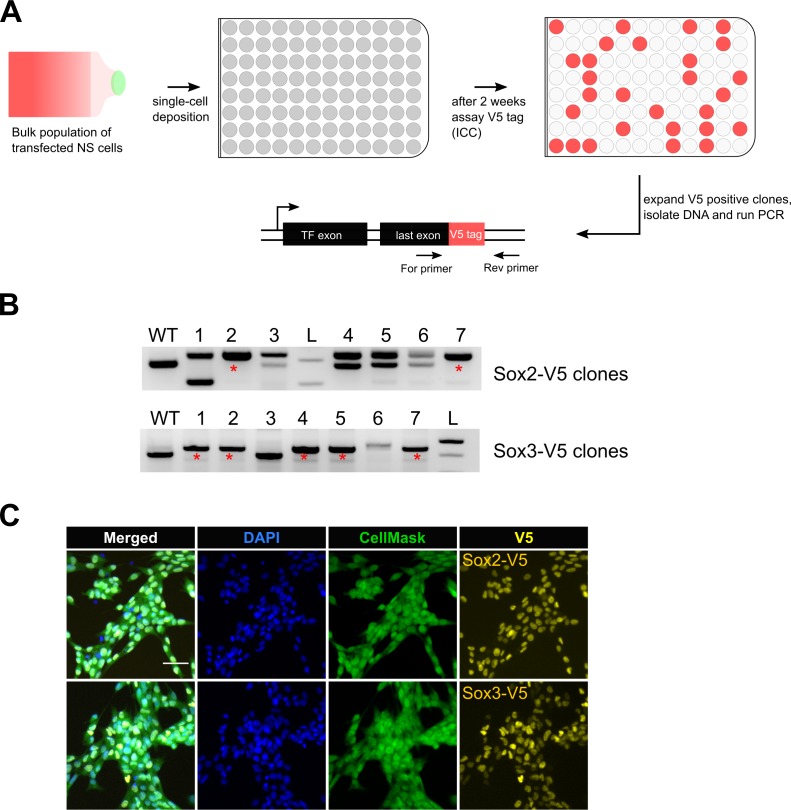
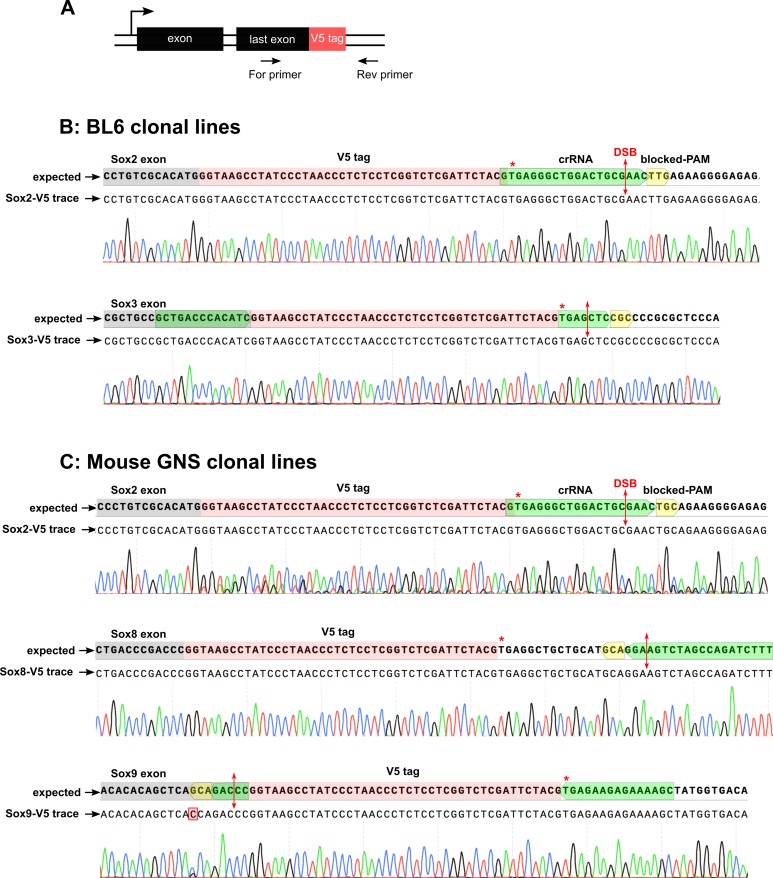
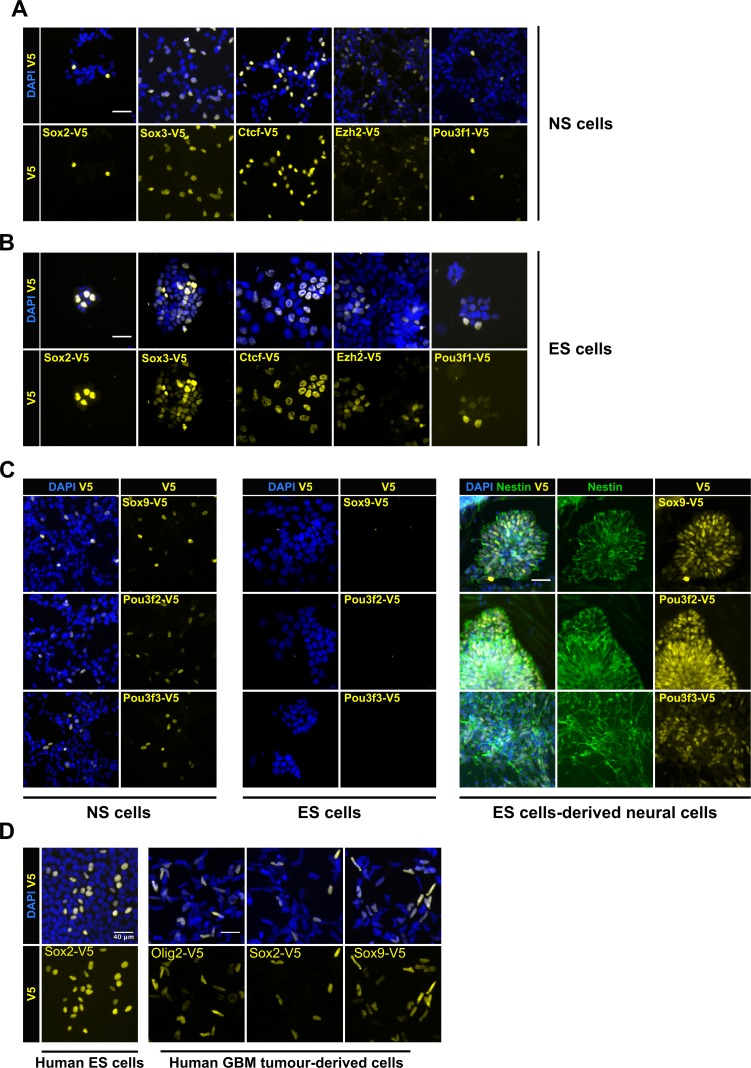
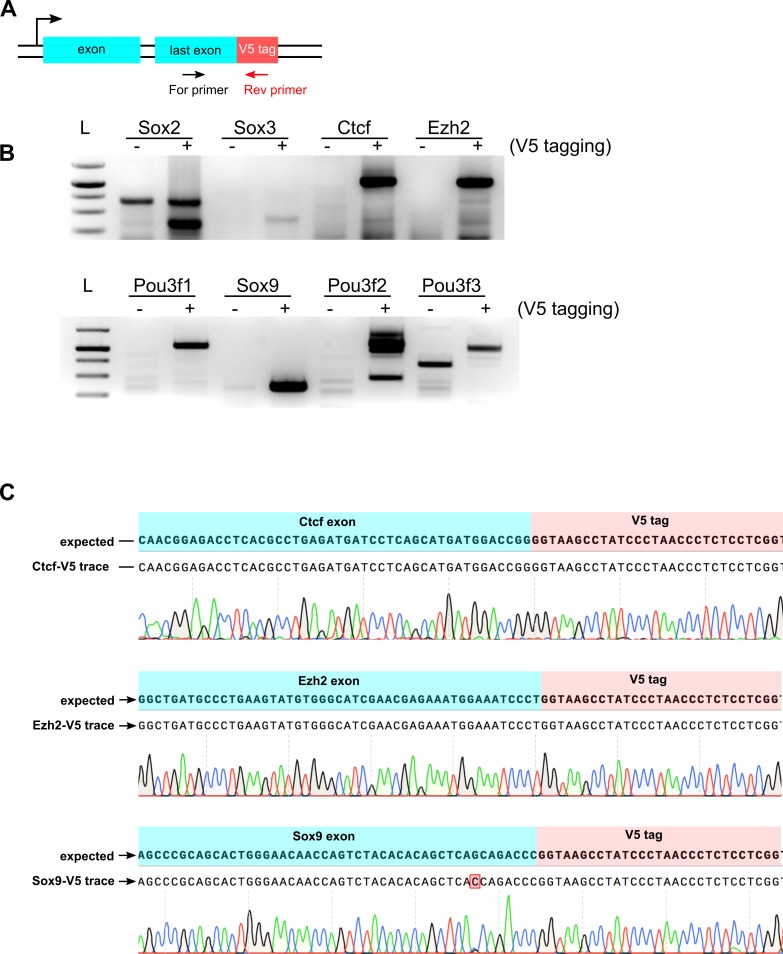
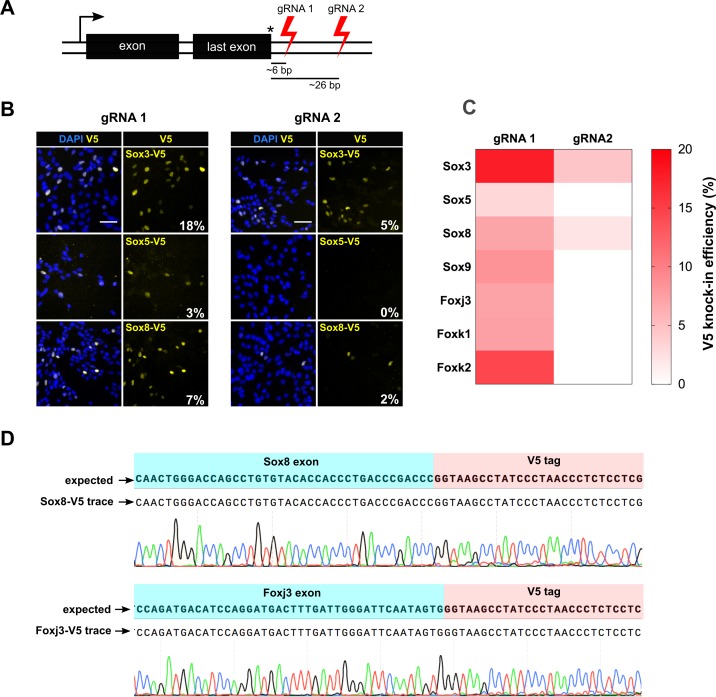
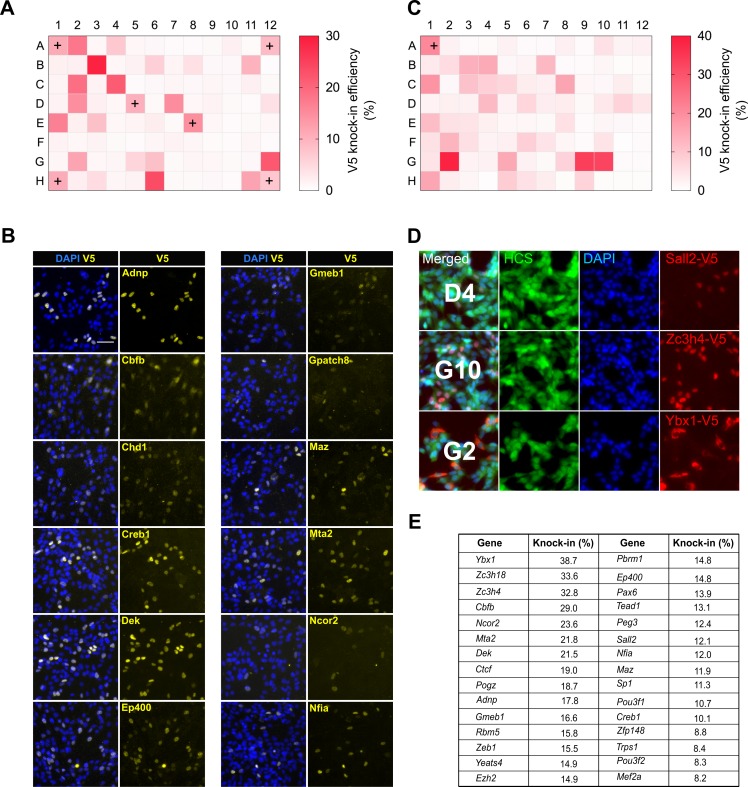
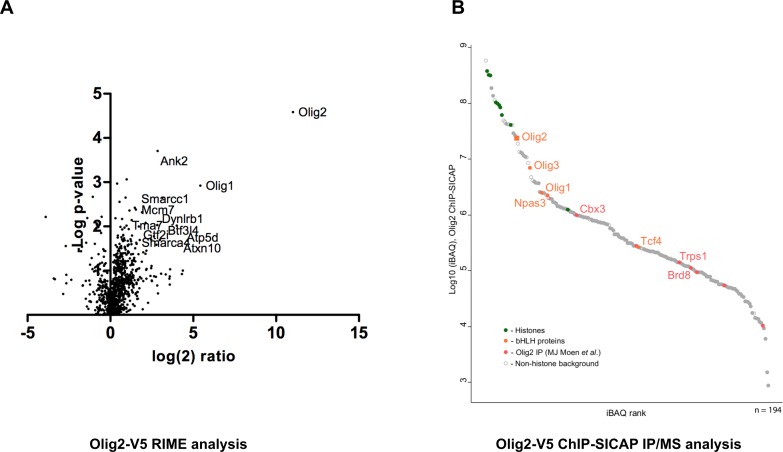
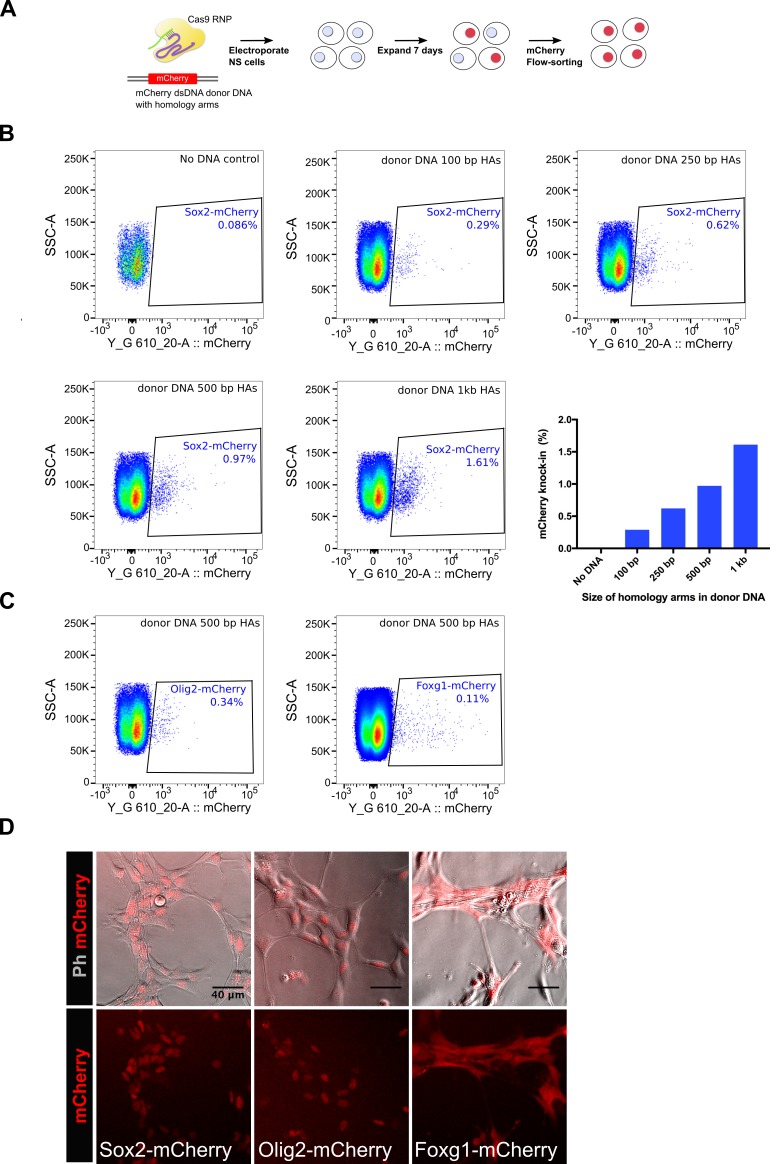
CRISPR/Cas9 can be used for precise genetic knock-in of epitope tags into endogenous genes, simplifying experimental analysis of protein function. However, Cas9-assisted epitope tagging in primary mammalian cell cultures is often inefficient and reliant on plasmid-based selection strategies. Here, we demonstrate improved knock-in efficiencies of diverse tags (V5, 3XFLAG, Myc, HA) using co-delivery of Cas9 protein pre-complexed with two-part synthetic modified RNAs (annealed crRNA:tracrRNA) and single-stranded oligodeoxynucleotide (ssODN) repair templates. Knock-in efficiencies of ~5-30%, were achieved without selection in embryonic stem (ES) cells, neural stem (NS) cells, and brain-tumor-derived stem cells. Biallelic-tagged clonal lines were readily derived and used to define Olig2 chromatin-bound interacting partners. Using our novel web-based design tool, we established a 96-well format pipeline that enabled V5-tagging of 60 different transcription factors. This efficient, selection-free and scalable epitope tagging pipeline enables systematic surveys of protein expression levels, subcellular localization, and interactors across diverse mammalian stem cells.
Keywords: CRISPR; cell biology; developmental biology; epitope tagging; genome editing; human; mouse; neural stem cell; stem cells; transcription factors.
© 2018, Dewari et al.
Conflict of interest statement
PD, BS, KM, GM, EO, NQ, AT, ML, CP, MK, CB, RF, RB, GM, Av, ST, JK, SP No competing interests declared, AJ employed by Integrated DNA Technologies (IDT), who sells reagents similar to some described herein. IDT is, however, not a publicly traded company and the authors do not own any shares or equity in IDT. No other authors have any financial interests or relationships with IDT; nor do they own any shares or equity, MB Mark A Behlke: employed by Integrated DNA Technologies (IDT), who sells reagents similar to some described herein. IDT is, however, not a publicly traded company and the authors do not own any shares or equity in IDT. No other authors have any financial interests or relationships with IDT; nor do they own any shares or equity
Figures
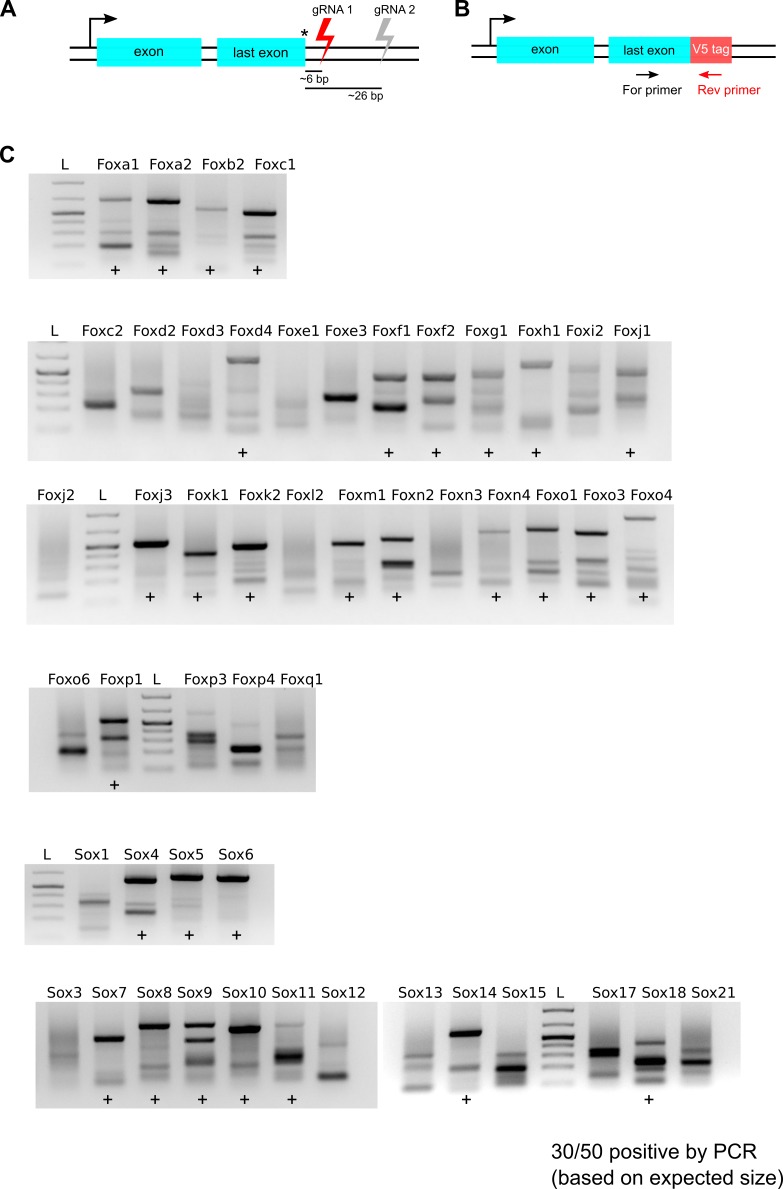
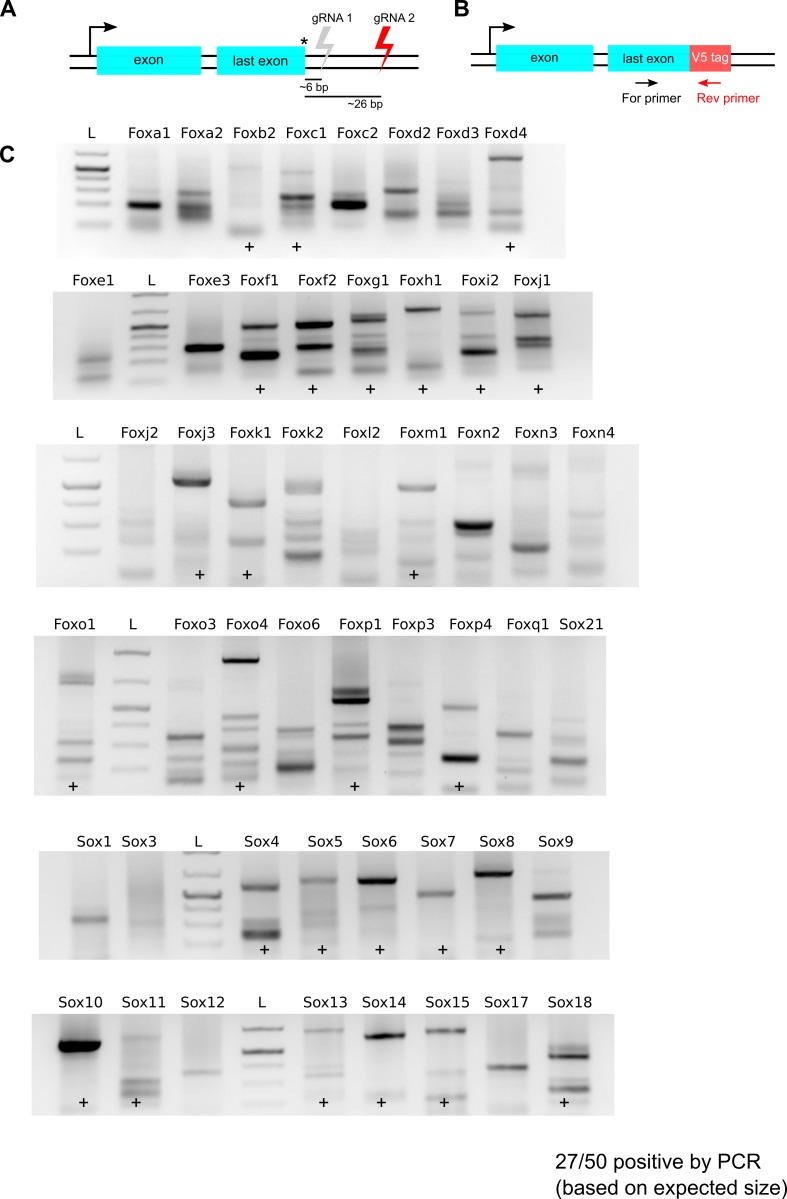
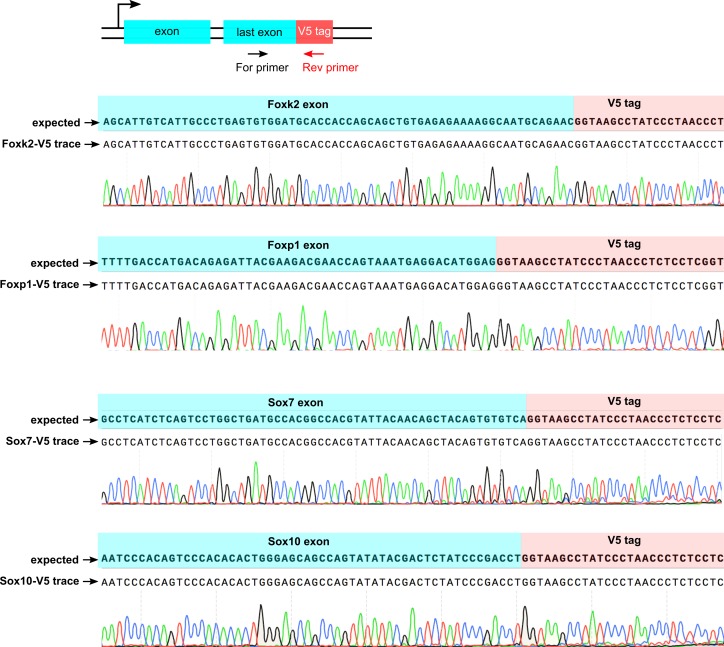
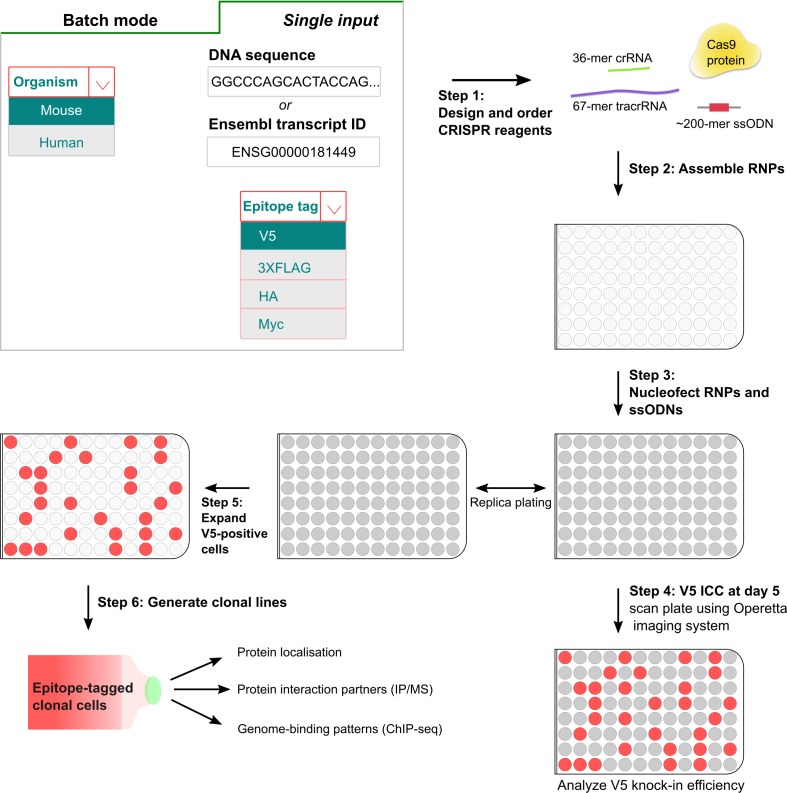
References
-
- Anderson EM, Haupt A, Schiel JA, Chou E, Machado HB, Strezoska Ž, Lenger S, McClelland S, Birmingham A, Vermeulen A, Smith A. Systematic analysis of CRISPR-Cas9 mismatch tolerance reveals low levels of off-target activity. Journal of Biotechnology. 2015;211:56–65. doi: 10.1016/j.jbiotec.2015.06.427. - DOI - PubMed
-
- Bressan RB, Dewari PS, Kalantzaki M, Gangoso E, Matjusaitis M, Garcia-Diaz C, Blin C, Grant V, Bulstrode H, Gogolok S, Skarnes WC, Pollard SM. Efficient CRISPR/Cas9-assisted gene targeting enables rapid and precise genetic manipulation of mammalian neural stem cells. Development. 2017;144:635–648. doi: 10.1242/dev.140855. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
