

Sickle Cell Anemia and Its Phenotypes
- PMID: 29641911
- PMCID: PMC7613509
- DOI: 10.1146/annurev-genom-083117-021320
Sickle Cell Anemia and Its Phenotypes
Abstract
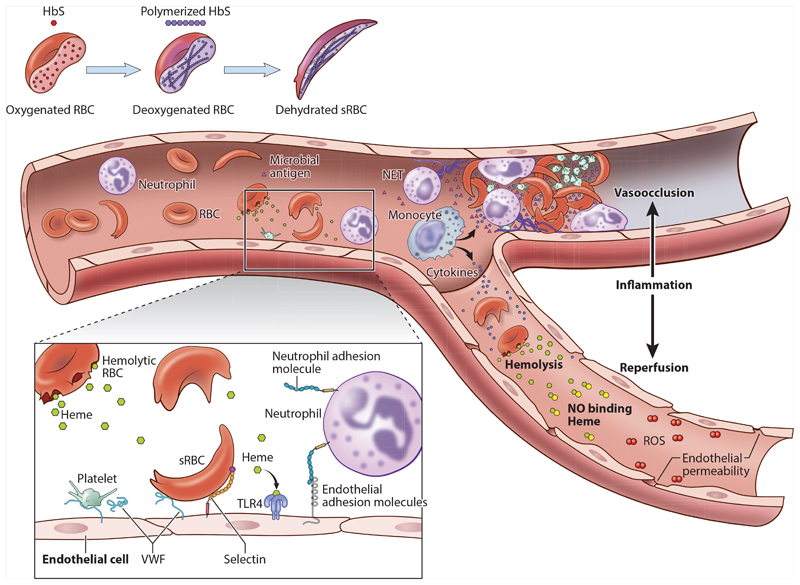
In the 100 years since sickle cell anemia (SCA) was first described in the medical literature, studies of its molecular and pathophysiological basis have been at the vanguard of scientific discovery. By contrast, the translation of such knowledge into treatments that improve the lives of those affected has been much too slow. Recent years, however, have seen major advances on several fronts. A more detailed understanding of the switch from fetal to adult hemoglobin and the identification of regulators such as BCL11A provide hope that these findings will be translated into genomic-based approaches to the therapeutic reactivation of hemoglobin F production in patients with SCA. Meanwhile, an unprecedented number of new drugs aimed at both the treatment and prevention of end-organ damage are now in the pipeline, outcomes from potentially curative treatments such as allogeneic hematopoietic stem cell transplantation are improving, and great strides are being made in gene therapy, where methods employing both antisickling β-globin lentiviral vectors and gene editing are now entering clinical trials. Encouragingly, after a century of neglect, the profile of the vast majority of those with SCA in Africa and India is also finally improving.
Keywords: Africa; genetic modifiers; genetics; genomics; sickle cell anemia.
Figures



References
-
- Abboud M, Laver J, Blau CA. Granulocytosis causing sickle-cell crisis. Lancet. 1998;351:959. - PubMed
-
- Adams R, McKie V, Nichols F, Carl E, Zhang D-L, et al. The use of transcranial ultrasonography to predict stroke in sickle cell disease. N Engl J Med. 1992;326:605–10. - PubMed
-
- Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5–11. - PubMed
-
- Adewoye AH, Nolan VG, Ma Q, Baldwin C, Wyszynski DF, et al. Association of polymorphisms of IGF1R and genes in the transforming growth factor–β/bone morphogenetic protein pathway with bacteremia in sickle cell anemia. Clin Infect Dis. 2006;43:593–98. - PubMed
-
- Adeyoju AB, Olujohungbe AB, Morris J, Yardumian A, Bareford D, et al. Priapism in sickle-cell disease; incidence, risk factors and complications – an international multicentre study. BJU Int. 2002;90:898–902. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
