

CYP51 as drug targets for fungi and protozoan parasites: past, present and future
- PMID: 29642960
- PMCID: PMC6185833
- DOI: 10.1017/S0031182018000562
CYP51 as drug targets for fungi and protozoan parasites: past, present and future
Abstract
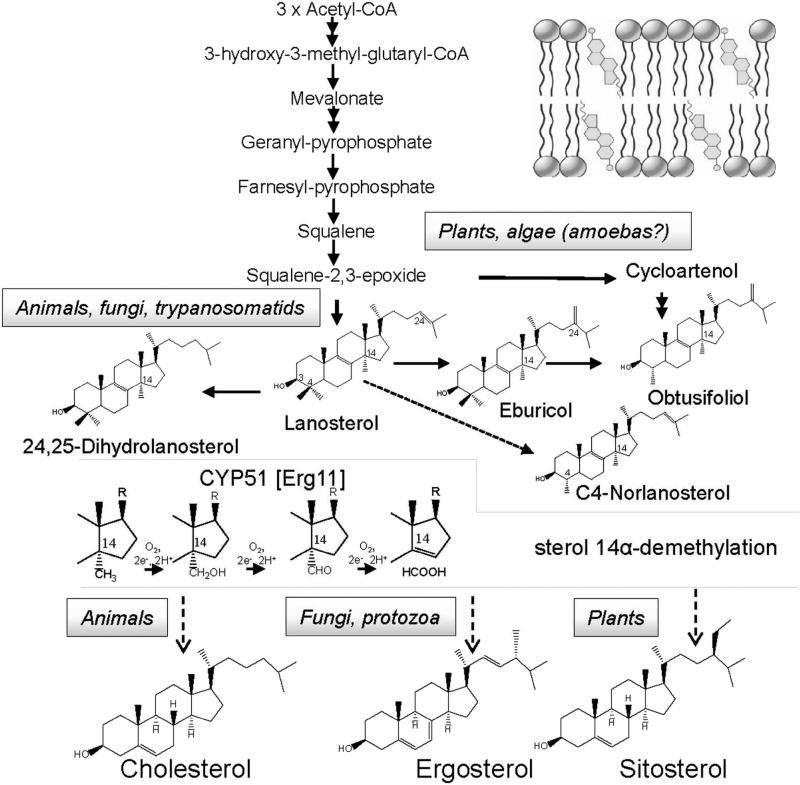
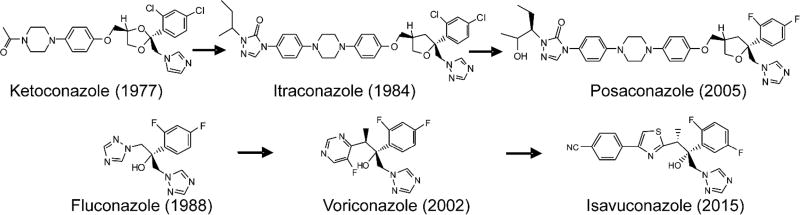
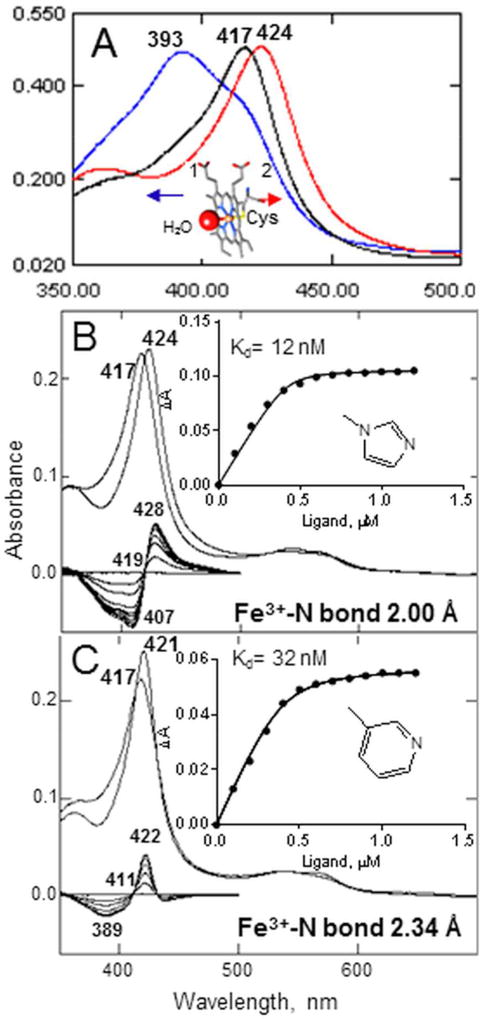
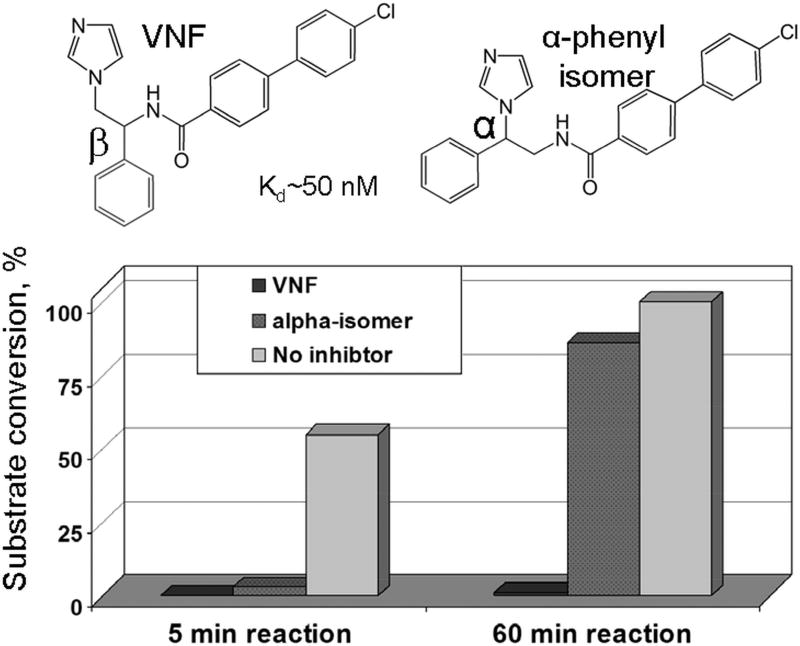
The efficiency of treatment of human infections with the unicellular eukaryotic pathogens such as fungi and protozoa remains deeply unsatisfactory. For example, the mortality rates from nosocomial fungemia in critically ill, immunosuppressed or post-cancer patients often exceed 50%. A set of six systemic clinical azoles [sterol 14α-demethylase (CYP51) inhibitors] represents the first-line antifungal treatment. All these drugs were discovered empirically, by monitoring their effects on fungal cell growth, though it had been proven that they kill fungal cells by blocking the biosynthesis of ergosterol in fungi at the stage of 14α-demethylation of the sterol nucleus. This review briefs the history of antifungal azoles, outlines the situation with the current clinical azole-based drugs, describes the attempts of their repurposing for treatment of human infections with the protozoan parasites that, similar to fungi, also produce endogenous sterols, and discusses the most recently acquired knowledge on the CYP51 structure/function and inhibition. It is our belief that this information should be helpful in shifting from the traditional phenotypic screening to the actual target-driven drug discovery paradigm, which will rationalize and substantially accelerate the development of new, more efficient and pathogen-oriented CYP51 inhibitors.
Keywords: Amoeba; CYP51; Leishmania; Trypanosoma brucei; Trypanosoma cruzi; antifungal azoles; crystal structure; inhibition; sterol 14α-demethylase; sterol biosynthesis.
Figures

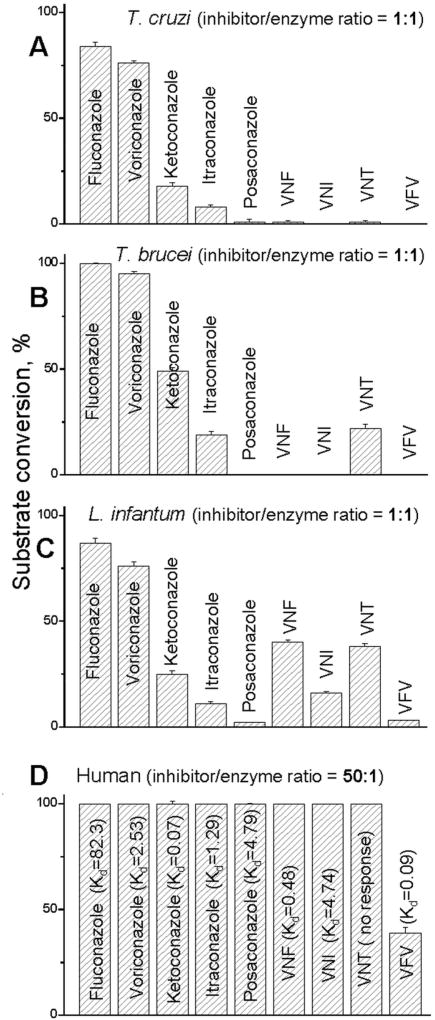
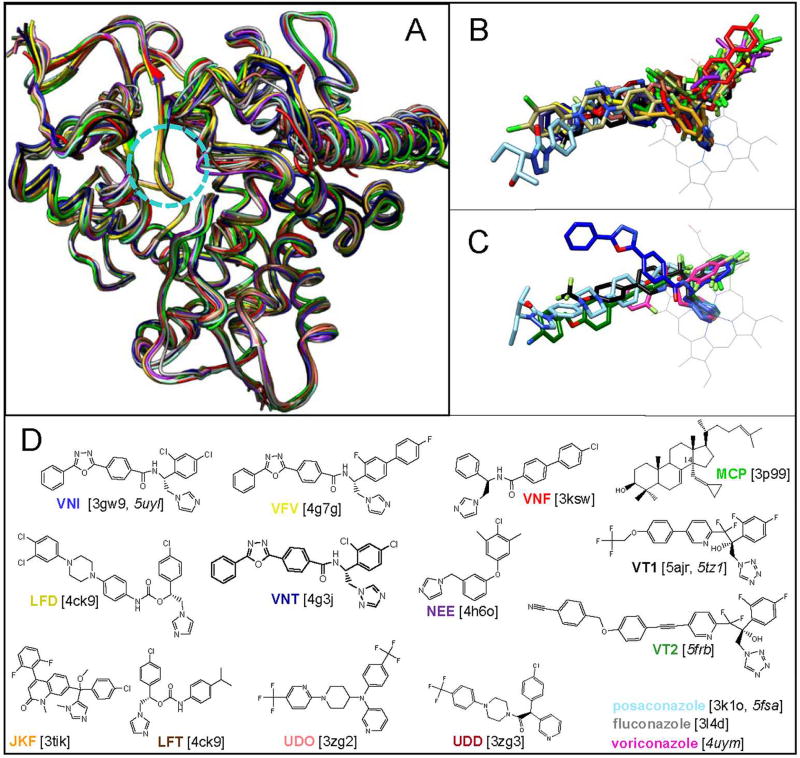
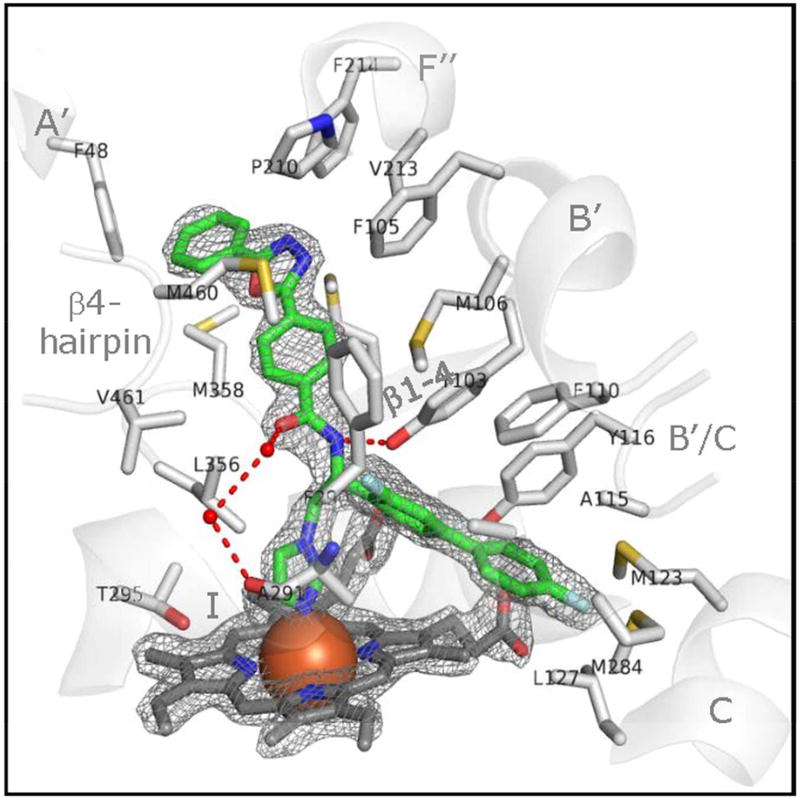
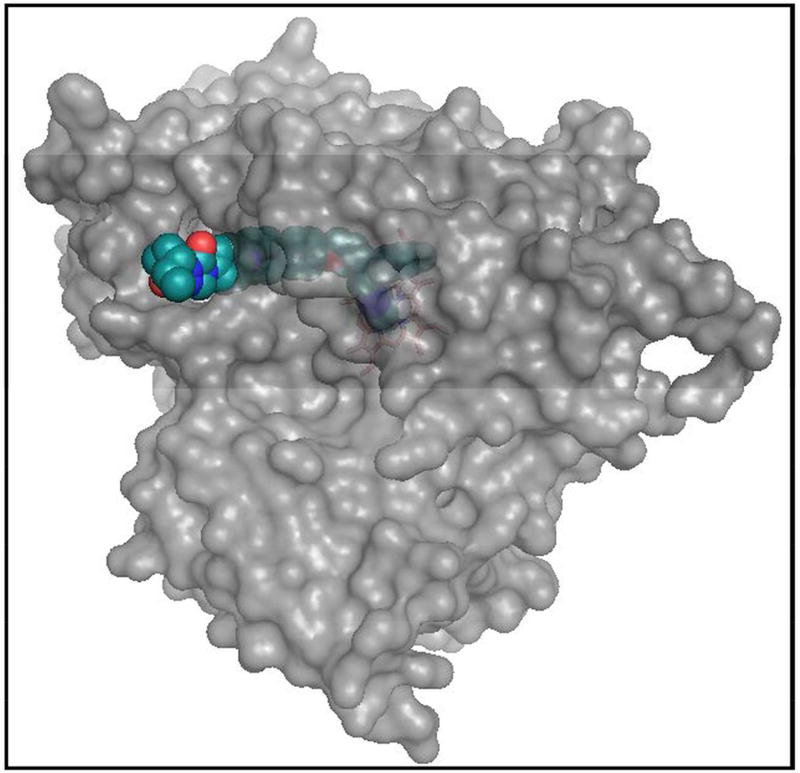
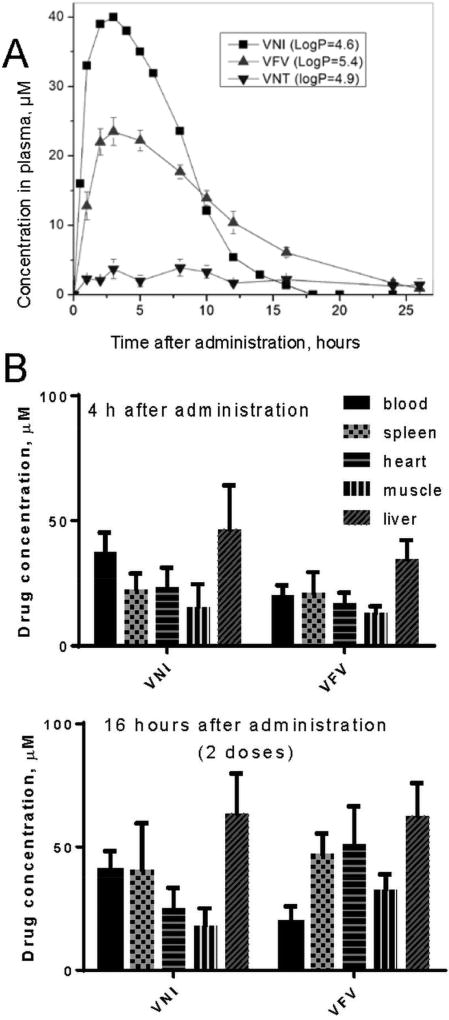
References
-
- Amoils SP, Heney C. Acanthamoeba keratitis with live isolates treated with cryosurgery and fluconazole. American Journal of Ophthalmology. 1999;127:718–720. - PubMed
-
- Andriani G, Amata E, Beatty J, Clements Z, Coffey BJ, Courtemanche G, Devine W, Erath J, Juda CE, Wawrzak Z, Wood JT, Lepesheva GI, Rodriguez A, Pollastri MP. Antitrypanosomal lead discovery: identification of a ligand-efficient inhibitor of Trypanosoma cruzi CYP51 and parasite growth. Journal of Medicinal Chemistry. 2013;56:2556–2567. doi: 10.1021/jm400012e. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
