

Contextual determinants of TGFβ action in development, immunity and cancer
- PMID: 29643418
- PMCID: PMC7457231
- DOI: 10.1038/s41580-018-0007-0
Contextual determinants of TGFβ action in development, immunity and cancer
Erratum in
-
Publisher Correction: Contextual determinants of TGFβ action in development, immunity and cancer.Nat Rev Mol Cell Biol. 2018 Jul;19(7):479. doi: 10.1038/s41580-018-0018-x. Nat Rev Mol Cell Biol. 2018. PMID: 29740128
Abstract
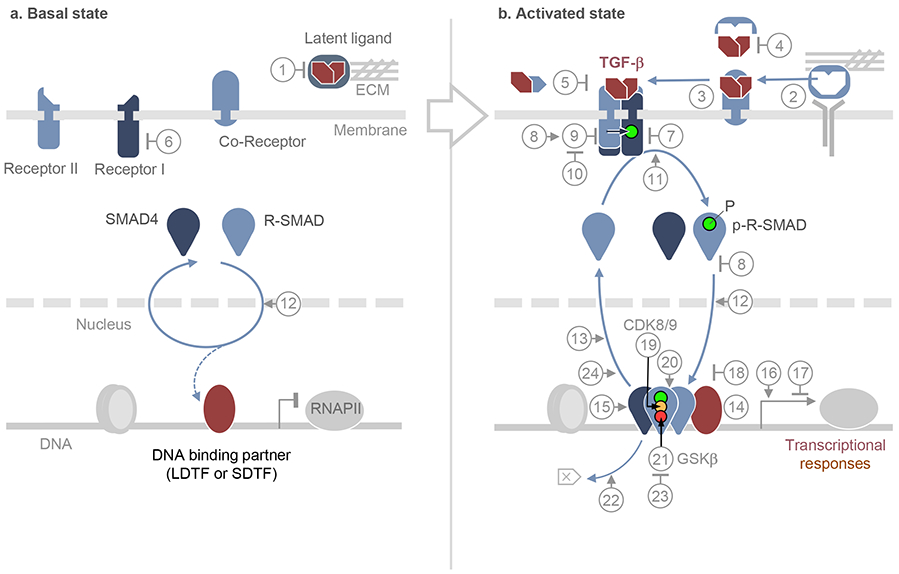
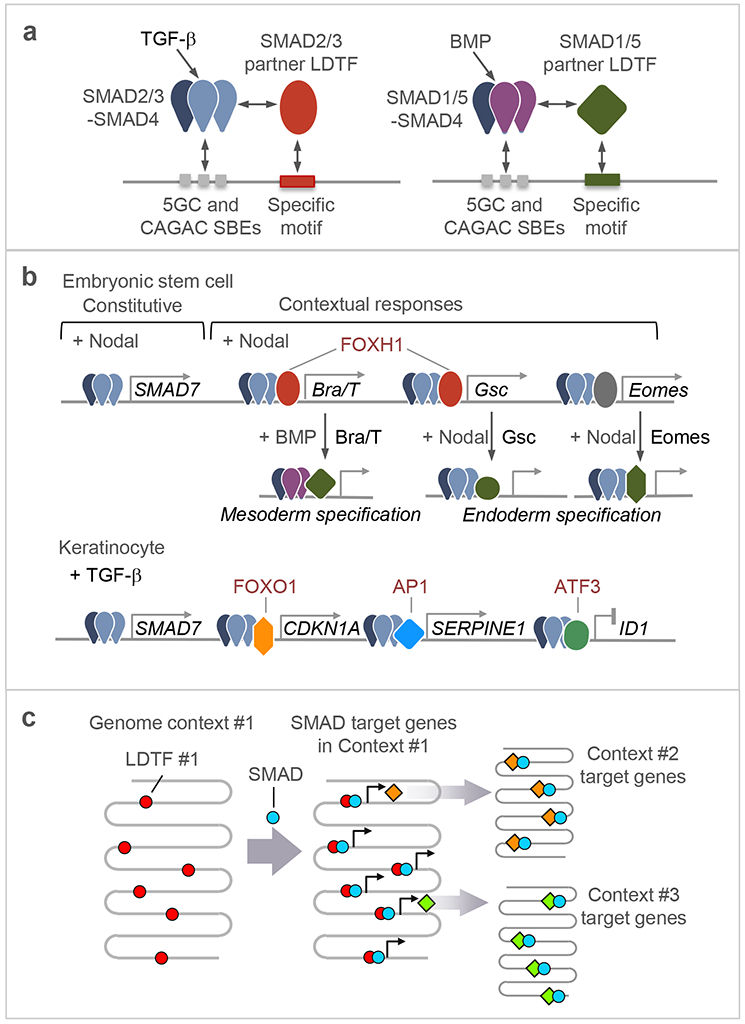
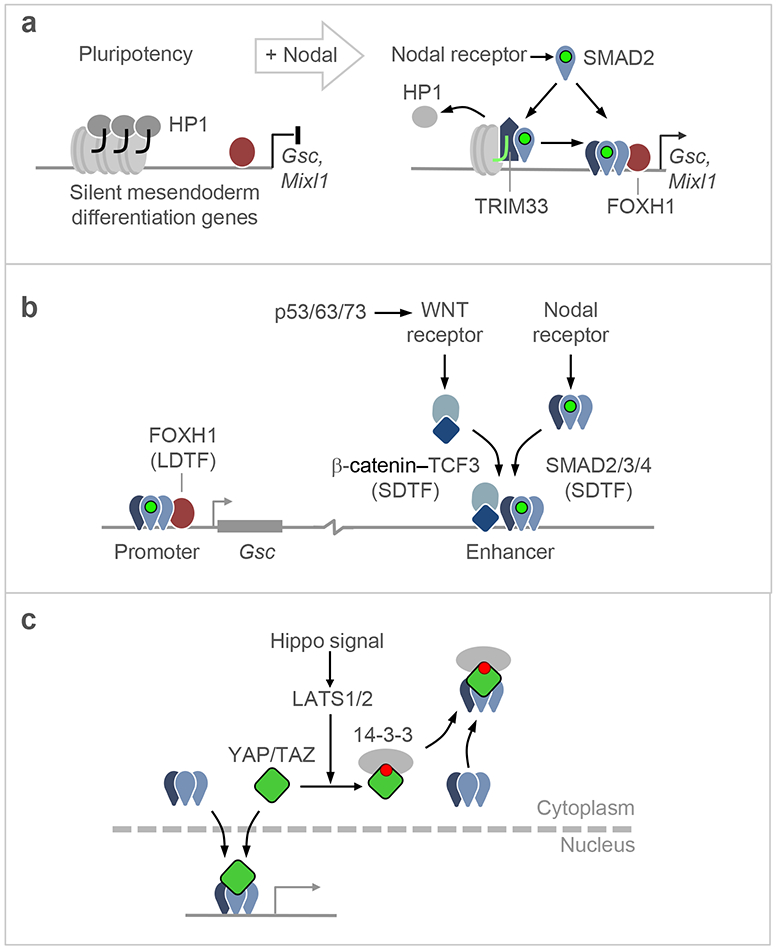
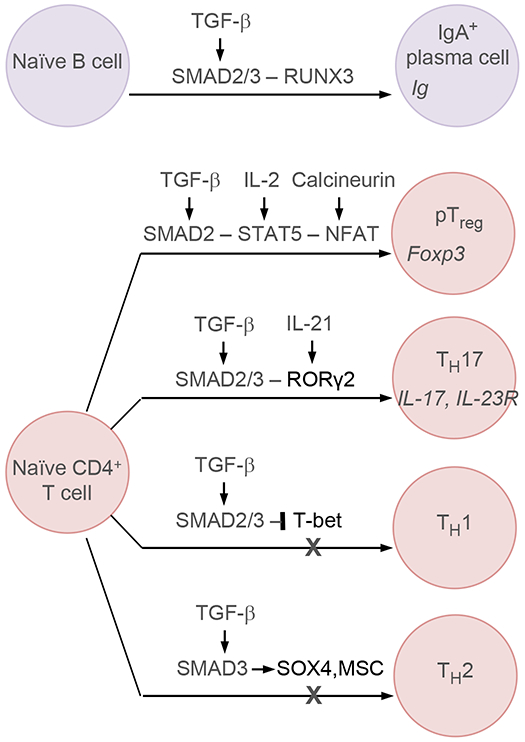
Few cell signals match the impact of the transforming growth factor-β (TGFβ) family in metazoan biology. TGFβ cytokines regulate cell fate decisions during development, tissue homeostasis and regeneration, and are major players in tumorigenesis, fibrotic disorders, immune malfunctions and various congenital diseases. The effects of the TGFβ family are mediated by a combinatorial set of ligands and receptors and by a common set of receptor-activated mothers against decapentaplegic homologue (SMAD) transcription factors, yet the effects can differ dramatically depending on the cell type and the conditions. Recent progress has illuminated a model of TGFβ action in which SMADs bind genome-wide in partnership with lineage-determining transcription factors and additionally integrate inputs from other pathways and the chromatin to trigger specific cellular responses. These new insights clarify the operating logic of the TGFβ pathway in physiology and disease.
Figures
References
-
- Massagué J How cells read TGF-beta signals. Nat Rev Mol Cell Biol 1, 169–178 (2000). - PubMed
-
- Josso N, Belville C, di Clemente N & Picard JY AMH and AMH receptor defects in persistent Mullerian duct syndrome. Hum Reprod Update 11, 351–356 (2005). - PubMed
-
- Matzuk MM & Burns KH Genetics of mammalian reproduction: modeling the end of the germline. Annu Rev Physiol 74, 503–528 (2012). - PubMed
-
- Meng XM, Nikolic-Paterson DJ & Lan HY TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol 12, 325–338 (2016). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
