

A hybrid reference-guided de novo assembly approach for generating Cyclospora mitochondrion genomes
- PMID: 29643938
- PMCID: PMC5891936
- DOI: 10.1186/s13099-018-0242-0
A hybrid reference-guided de novo assembly approach for generating Cyclospora mitochondrion genomes
Abstract
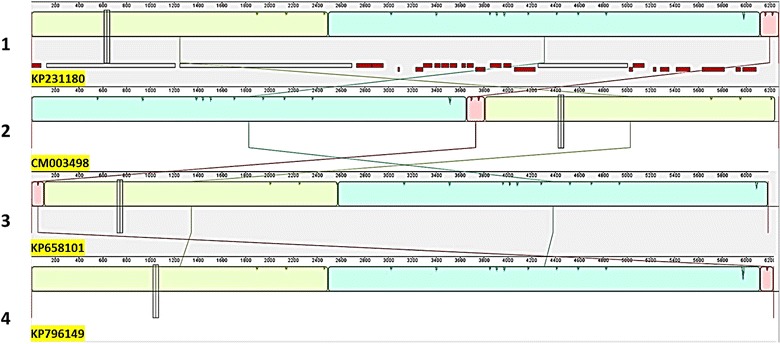
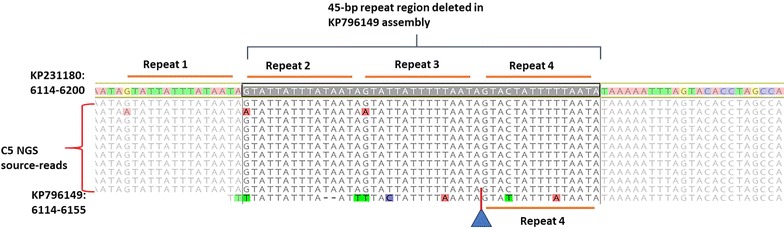
Cyclospora cayetanensis is a coccidian parasite associated with large and complex foodborne outbreaks worldwide. Linking samples from cyclosporiasis patients during foodborne outbreaks with suspected contaminated food sources, using conventional epidemiological methods, has been a persistent challenge. To address this issue, development of new methods based on potential genomically-derived markers for strain-level identification has been a priority for the food safety research community. The absence of reference genomes to identify nucleotide and structural variants with a high degree of confidence has limited the application of using sequencing data for source tracking during outbreak investigations. In this work, we determined the quality of a high resolution, curated, public mitochondrial genome assembly to be used as a reference genome by applying bioinformatic analyses. Using this reference genome, three new mitochondrial genome assemblies were built starting with metagenomic reads generated by sequencing DNA extracted from oocysts present in stool samples from cyclosporiasis patients. Nucleotide variants were identified in the new and other publicly available genomes in comparison with the mitochondrial reference genome. A consolidated workflow, presented here, to generate new mitochondrion genomes using our reference-guided de novo assembly approach could be useful in facilitating the generation of other mitochondrion sequences, and in their application for subtyping C. cayetanensis strains during foodborne outbreak investigations.
Keywords: Cyclosporiasis; De novo assembly; Genome sequencing; Mitochondrion; Reference genome; Single nucleotide polymorphisms; Subtyping.
Figures


References
-
- Chacin-Bonilla, L. 2017. Cyclospora cayetanensis. In: JB Rose and B Jiménez-Cisneros, editors. Global water pathogens project. http://www.waterpathogens.org (R.Fayer and W. Jakubowski, editor Part 3 Protists). http://www.waterpathogens.org/book/cyclospora-cayetanensis. E. Lansing: Michigan State University, UNESCO.
-
- Cinar HN, Qvarnstrom Y, Wei-Pridgeon Y, Li W, Nascimento FS, Arrowood MJ, Murphy HR, Jang A, Kim E, Kim R, da Silva A, Gopinath GR. Comparative sequence analysis of Cyclospora cayetanensis apicoplast genomes originating from diverse geographical regions. Parasit Vectors. 2016;9(1):611. doi: 10.1186/s13071-016-1896-4. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
