

Regulation of Blood Pressure by Targeting CaV1.2-Galectin-1 Protein Interaction
- PMID: 29650545
- PMCID: PMC6185826
- DOI: 10.1161/CIRCULATIONAHA.117.031231
Regulation of Blood Pressure by Targeting CaV1.2-Galectin-1 Protein Interaction
Abstract
Background: L-type CaV1.2 channels play crucial roles in the regulation of blood pressure. Galectin-1 (Gal-1) has been reported to bind to the I-II loop of CaV1.2 channels to reduce their current density. However, the mechanistic understanding for the downregulation of CaV1.2 channels by Gal-1 and whether Gal-1 plays a direct role in blood pressure regulation remain unclear.
Methods: In vitro experiments involving coimmunoprecipitation, Western blot, patch-clamp recordings, immunohistochemistry, and pressure myography were used to evaluate the molecular mechanisms by which Gal-1 downregulates CaV1.2 channel in transfected, human embryonic kidney 293 cells, smooth muscle cells, arteries from Lgasl1-/- mice, rat, and human patients. In vivo experiments involving the delivery of Tat-e9c peptide and AAV5-Gal-1 into rats were performed to investigate the effect of targeting CaV1.2-Gal-1 interaction on blood pressure monitored by tail-cuff or telemetry methods.
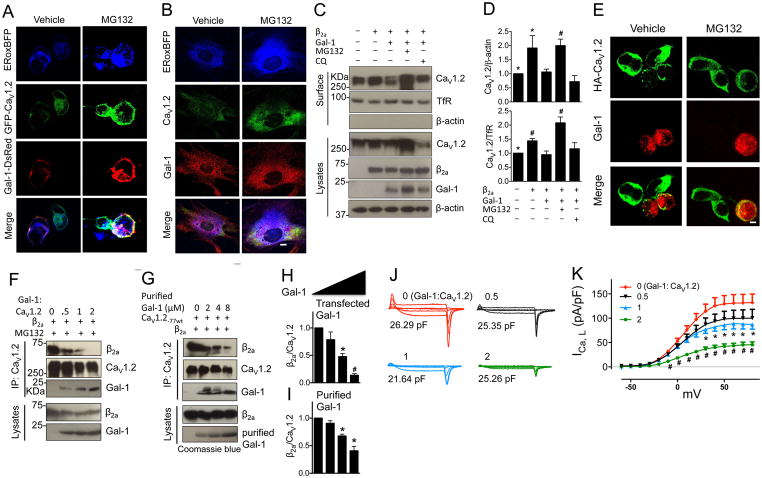
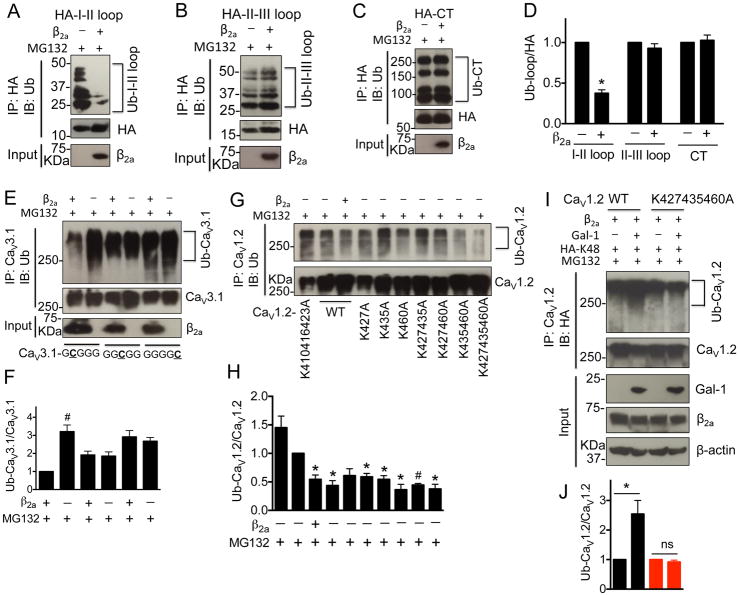
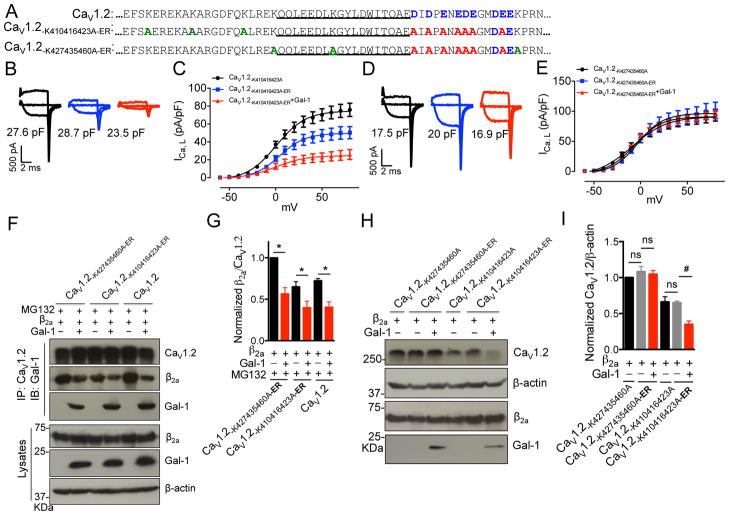
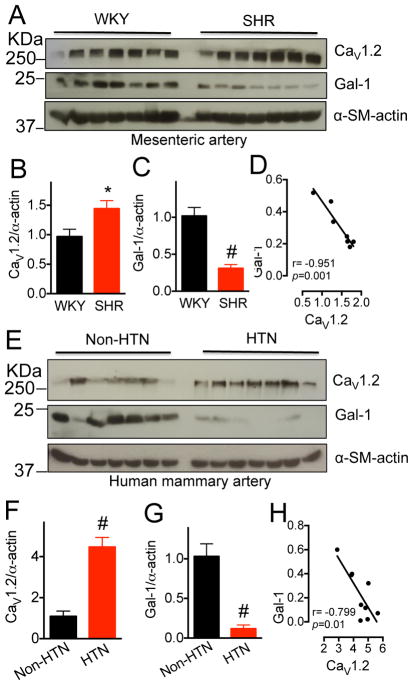
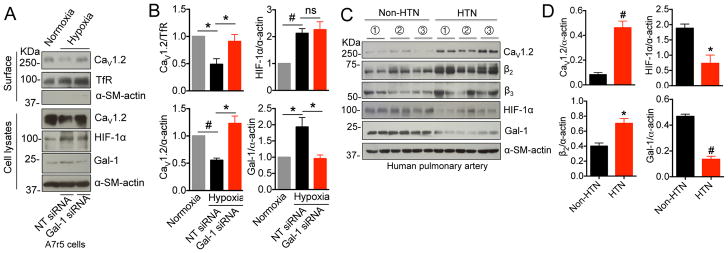
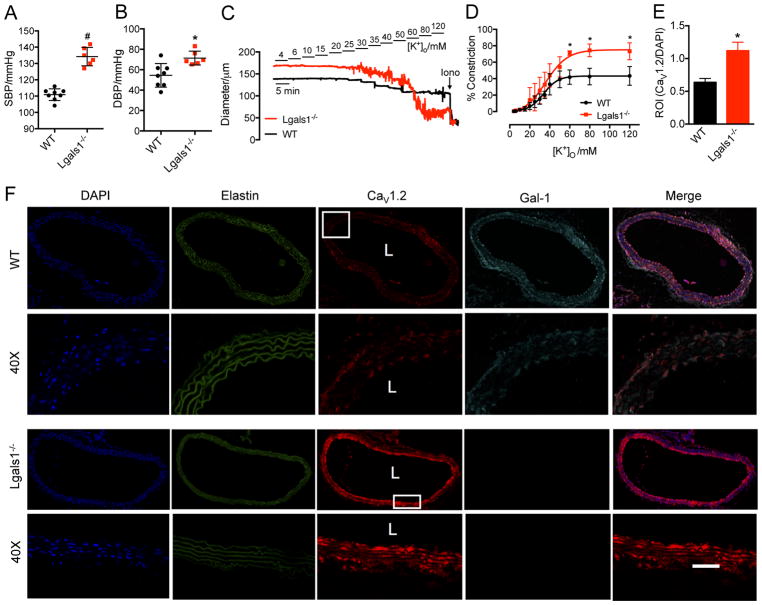
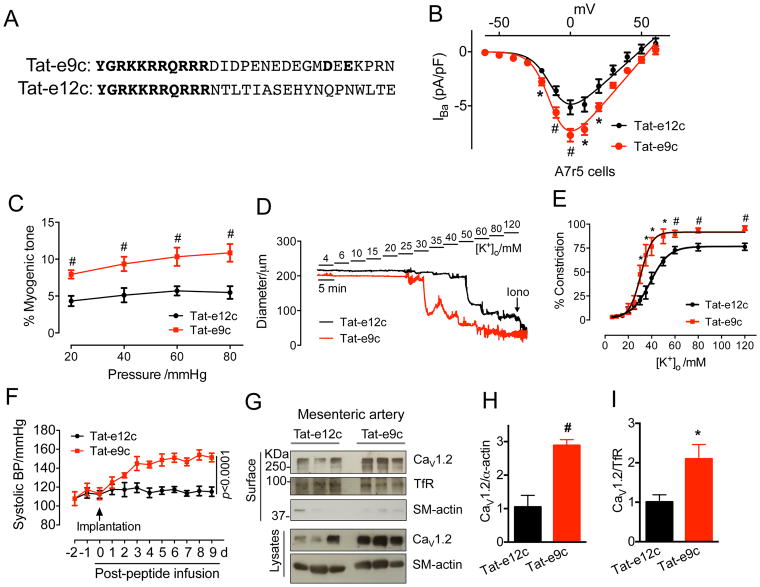
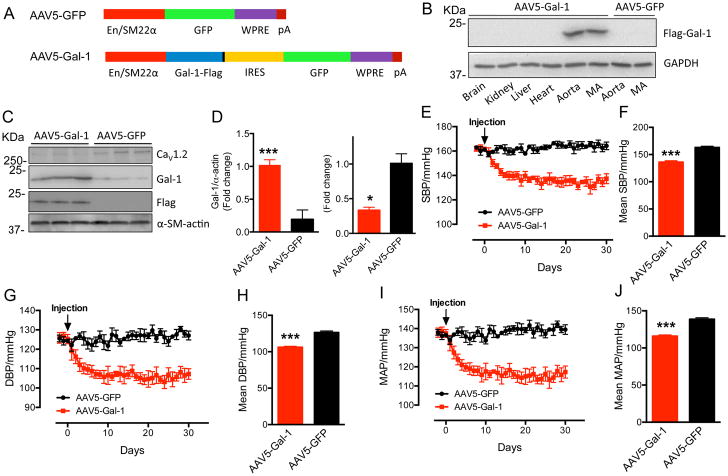
Results: Our study reveals that Gal-1 is a key regulator for proteasomal degradation of CaV1.2 channels. Gal-1 competed allosterically with the CaVβ subunit for binding to the I-II loop of the CaV1.2 channel. This competitive disruption of CaVβ binding led to CaV1.2 degradation by exposing the channels to polyubiquitination. It is notable that we demonstrated that the inverse relationship of reduced Gal-1 and increased CaV1.2 protein levels in arteries was associated with hypertension in hypertensive rats and patients, and Gal-1 deficiency induces higher blood pressure in mice because of the upregulated CaV1.2 protein level in arteries. To directly regulate blood pressure by targeting the CaV1.2-Gal-1 interaction, we administered Tat-e9c, a peptide that competed for binding of Gal-1 by a miniosmotic pump, and this specific disruption of CaV1.2-Gal-1 coupling increased smooth muscle CaV1.2 currents, induced larger arterial contraction, and caused hypertension in rats. In contrasting experiments, overexpression of Gal-1 in smooth muscle by a single bolus of AAV5-Gal-1 significantly reduced blood pressure in spontaneously hypertensive rats.
Conclusions: We have defined molecularly that Gal-1 promotes CaV1.2 degradation by replacing CaVβ and thereby exposing specific lysines for polyubiquitination and by masking I-II loop endoplasmic reticulum export signals. This mechanistic understanding provided the basis for targeting CaV1.2-Gal-1 interaction to demonstrate clearly the modulatory role that Gal-1 plays in regulating blood pressure, and offering a potential approach for therapeutic management of hypertension.
Keywords: blood pressure; calcium channels, L type; galectin-1; hypertension; proteasome endopeptidase complex.
Figures
References
-
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB. Executive summary: Heart disease and stroke statistics--2016 update: A report from the american heart association. Circulation. 2016;133:447–454. - PubMed
-
- Rensen SS, Niessen PM, van Deursen JM, Janssen BJ, Heijman E, Hermeling E, Meens M, Lie N, Gijbels MJ, Strijkers GJ, Doevendans PA, Hofker MH, De Mey JG, van Eys GJ. Smoothelin-B deficiency results in reduced arterial contractility, hypertension, and cardiac hypertrophy in mice. Circulation. 2008;118:828–836. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
