

Identification of rare sequence variation underlying heritable pulmonary arterial hypertension
- PMID: 29650961
- PMCID: PMC5897357
- DOI: 10.1038/s41467-018-03672-4
Identification of rare sequence variation underlying heritable pulmonary arterial hypertension
Abstract
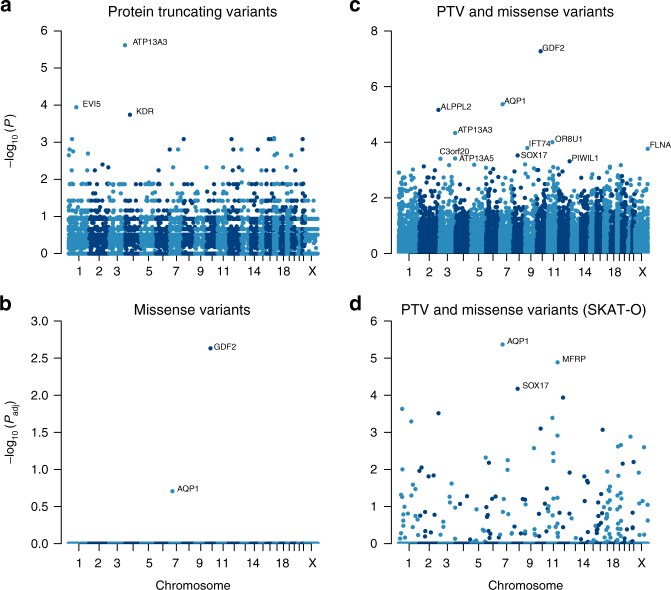
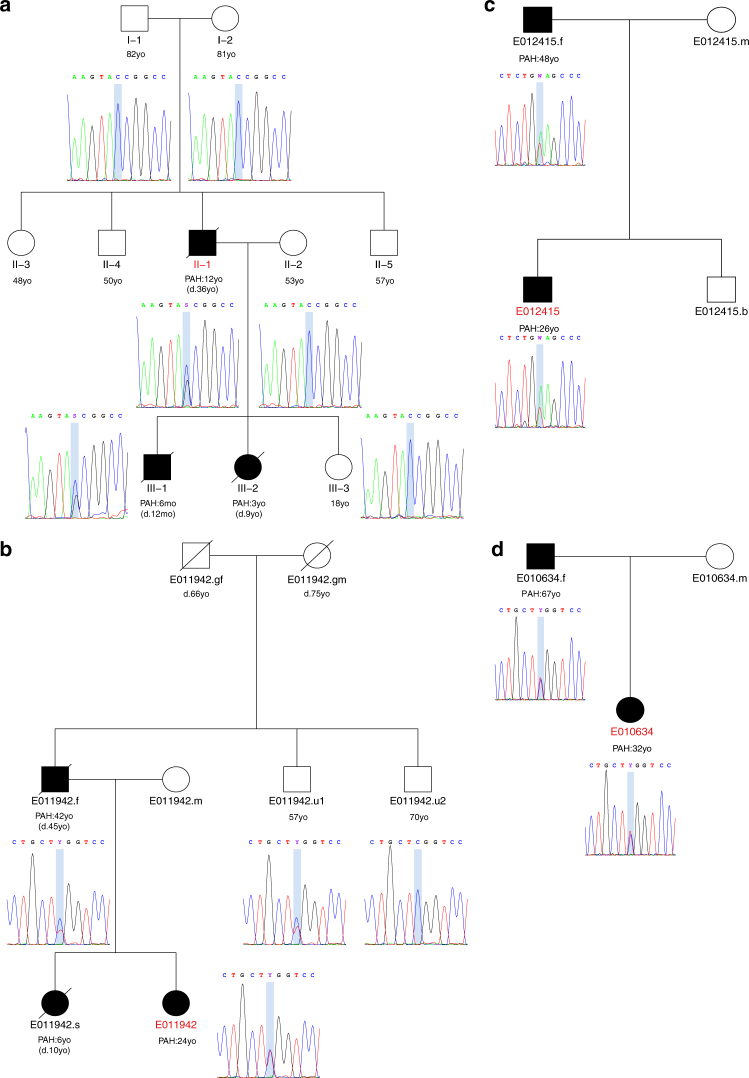
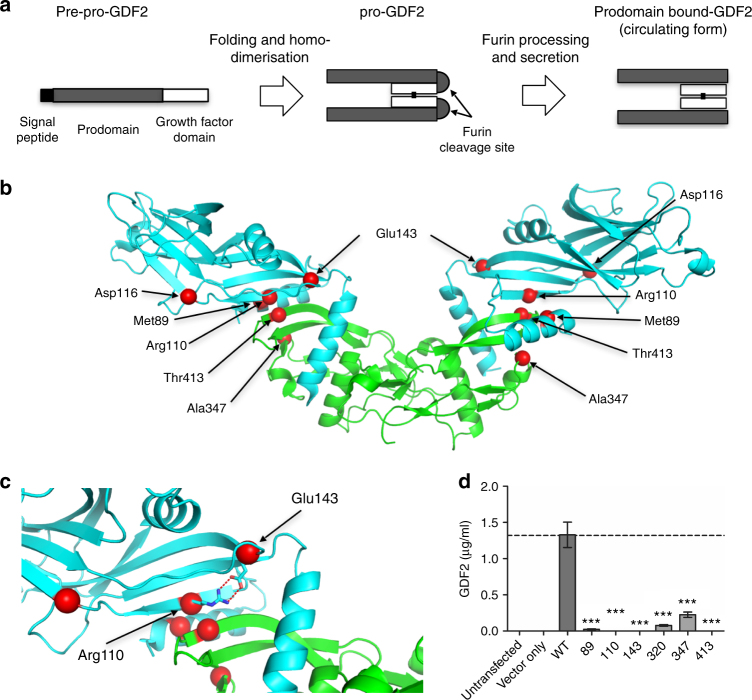
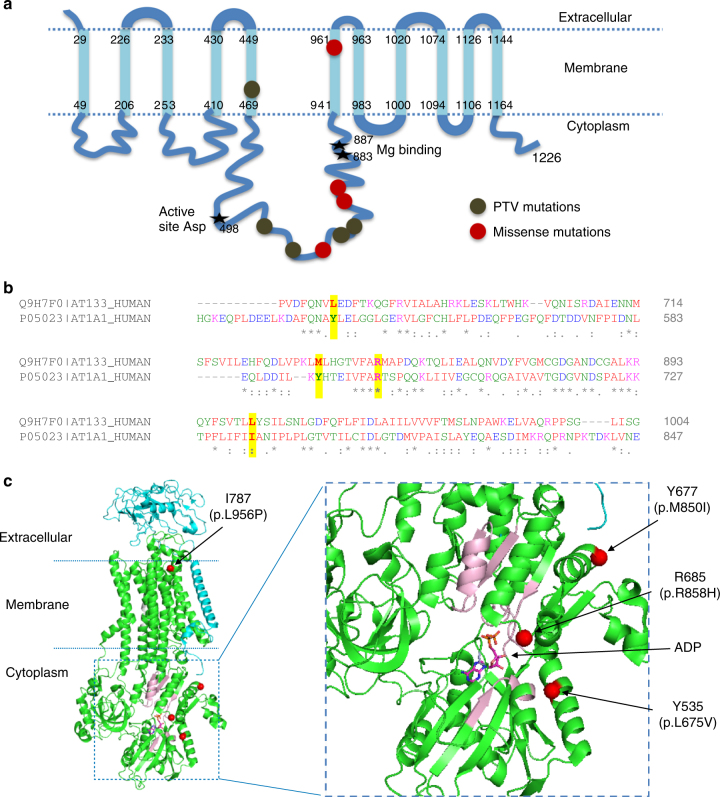
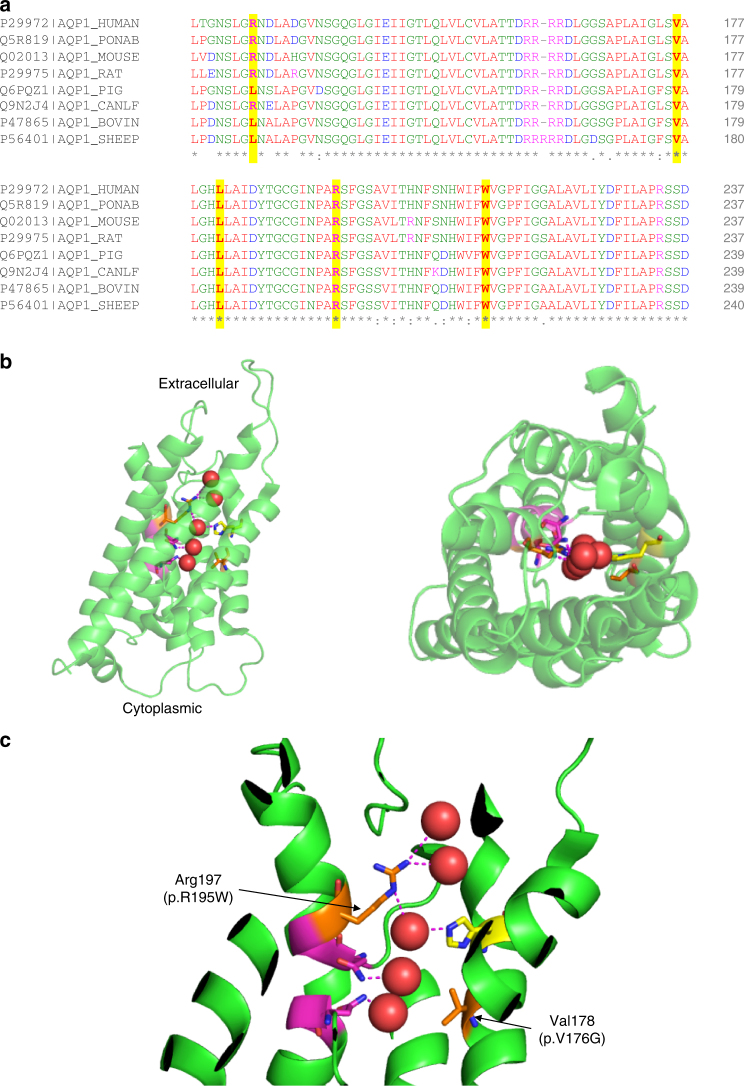
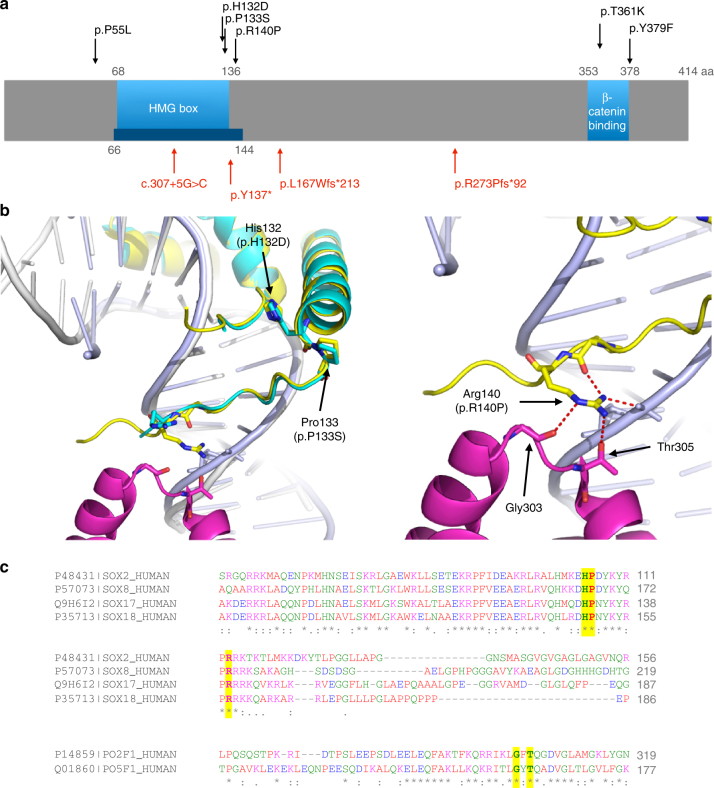
Pulmonary arterial hypertension (PAH) is a rare disorder with a poor prognosis. Deleterious variation within components of the transforming growth factor-β pathway, particularly the bone morphogenetic protein type 2 receptor (BMPR2), underlies most heritable forms of PAH. To identify the missing heritability we perform whole-genome sequencing in 1038 PAH index cases and 6385 PAH-negative control subjects. Case-control analyses reveal significant overrepresentation of rare variants in ATP13A3, AQP1 and SOX17, and provide independent validation of a critical role for GDF2 in PAH. We demonstrate familial segregation of mutations in SOX17 and AQP1 with PAH. Mutations in GDF2, encoding a BMPR2 ligand, lead to reduced secretion from transfected cells. In addition, we identify pathogenic mutations in the majority of previously reported PAH genes, and provide evidence for further putative genes. Taken together these findings contribute new insights into the molecular basis of PAH and indicate unexplored pathways for therapeutic intervention.
Conflict of interest statement
The authors declare no competing interests.
Figures
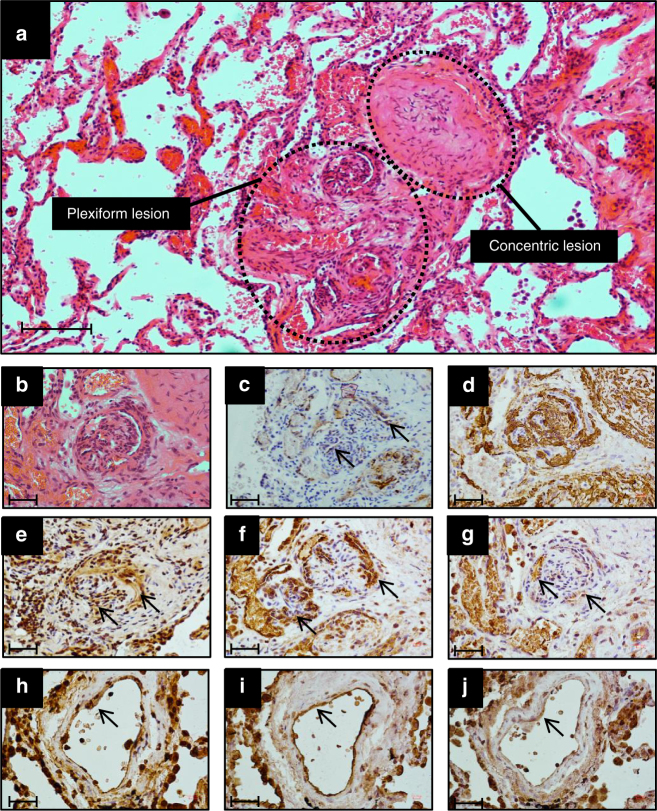
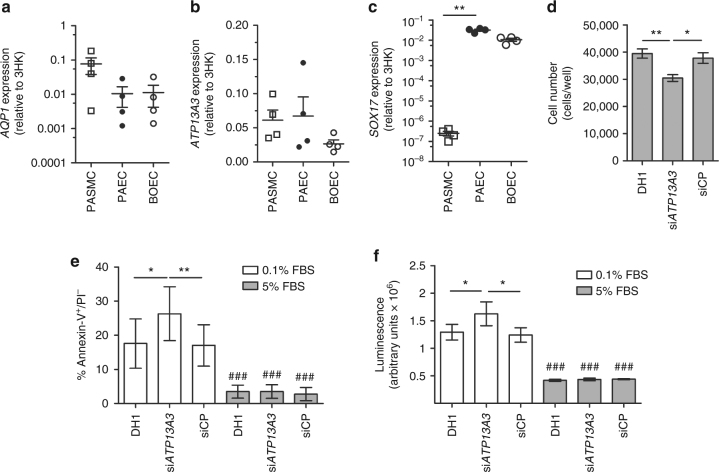
References
-
- Wagenvoort C. A. The pathology of primary pulmonary hypertension. J. Pathol.101, Pi (1970) - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- FS/15/59/31839/BHF_/British Heart Foundation/United Kingdom
- SP/12/12/29836/BHF_/British Heart Foundation/United Kingdom
- MR/K020919/1/MRC_/Medical Research Council/United Kingdom
- FS/13/48/30453/BHF_/British Heart Foundation/United Kingdom
- R01 HL098199/HL/NHLBI NIH HHS/United States
- PG/17/1/32532/BHF_/British Heart Foundation/United Kingdom
- PG/17/58/33134/BHF_/British Heart Foundation/United Kingdom
- WT_/Wellcome Trust/United Kingdom
- FS/18/52/33808/BHF_/British Heart Foundation/United Kingdom
- PG/15/39/31519/BHF_/British Heart Foundation/United Kingdom
- RG/13/4/30107/BHF_/British Heart Foundation/United Kingdom
- PG/12/54/29734/BHF_/British Heart Foundation/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
