

Inflammatory Events Following Subarachnoid Hemorrhage (SAH)
- PMID: 29651951
- PMCID: PMC6251050
- DOI: 10.2174/1570159X16666180412110919
Inflammatory Events Following Subarachnoid Hemorrhage (SAH)
Abstract
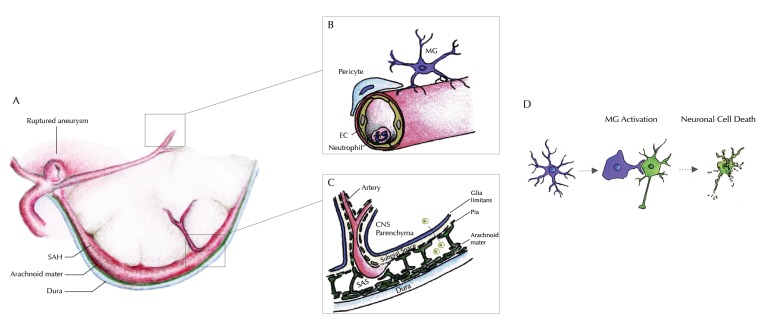
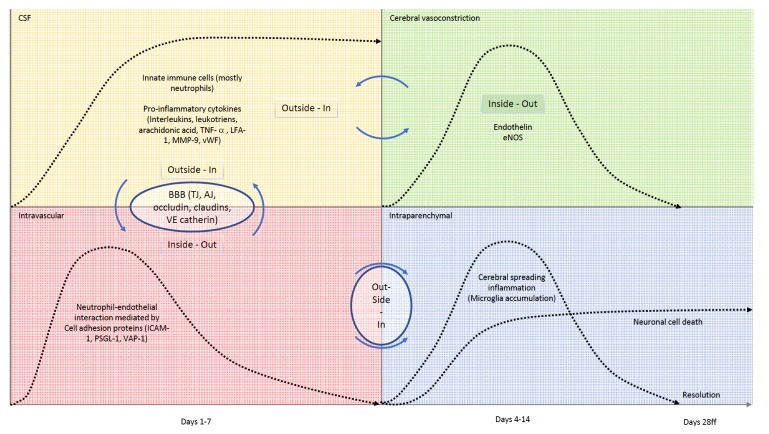
Acute SAH from a ruptured intracranial aneurysm contributes for 30% of all hemorrhagic strokes. The bleeding itself occurs in the subarachnoid space. Nevertheless, injury to the brain parenchyma occurs as a consequence of the bleeding, directly, via several well-defined mechanisms and pathways, but also indirectly, or secondarily. This secondary brain injury following SAH has a variety of causes and possible mechanisms. Amongst others, inflammatory events have been shown to occur in parallel to, contribute to, or even to initiate programmed cell death (PCD) within the central nervous system (CNS) in human and animal studies alike. Mechanisms of secondary brain injury are of utmost interest not only to scientists, but also to clinicians, as they often provide possibilities for translational approaches as well as distinct time windows for tailored treatment options. In this article, we review secondary brain injury due to inflammatory changes, that occur on cellular, as well as on molecular level in the various different compartments of the CNS: the brain vessels, the subarachnoid space, and the brain parenchyma itself and hypothesize about possible signaling mechanisms between these compartments.
Keywords: Subarachnoid hemorrhage; inflammation; microglia; neuronal cell death; secondary brain injury; secondary brain injury..
Copyright© Bentham Science Publishers; For any queries, please email at epub@benthamscience.org.
Figures
References
-
- Mathiesen T., Andersson B., Loftenius A. Holst, von. H. J. Neurosurg. 1993;78(4):562–567. [http://dx.doi.org/10.3171/jns.1993. 78.4.0562]. - PubMed
-
- Mathiesen T., Edner G., Ulfarsson E., Andersson B. Cerebrospinal fluid interleukin-1 receptor antagonist and tumor necrosis factor-alpha following subarachnoid hemorrhage. J. Neurosurg. 1997;87(2):215–220. [http://dx.doi.org/10.3171/jns.1997.87.2.0215]. [PMID: 9254084]. - PubMed
-
- Mathiesen T., Lefvert A.K. Cerebrospinal fluid and blood lymphocyte subpopulations following subarachnoid haemorrhage. Br. J. Neurosurg. 1996;10(1):89–92. [http://dx.doi.org/10.1080/ 02688699650040584]. [PMID: 8672265]. - PubMed
-
- Minami N., Tani E., Maeda Y., Yamaura I., Fukami M. Effects of inhibitors of protein kinase C and calpain in experimental delayed cerebral vasospasm. J. Neurosurg. 1992;76(1):111–118. [http://dx.doi.org/10.3171/jns.1992.76.1.0111]. [PMID: 1370069]. - PubMed
-
- Yoshimoto Y., Tanaka Y., Hoya K. Acute systemic inflammatory response syndrome in subarachnoid hemorrhage. Stroke. 2001;32(9):1989–1993. [http://dx.doi.org/10.1161/hs0901.095646]. [PMID: 11546886]. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources