

Cholesterol and bile acid-mediated regulation of autophagy in fatty liver diseases and atherosclerosis
- PMID: 29653253
- PMCID: PMC6037329
- DOI: 10.1016/j.bbalip.2018.04.005
Cholesterol and bile acid-mediated regulation of autophagy in fatty liver diseases and atherosclerosis
Abstract
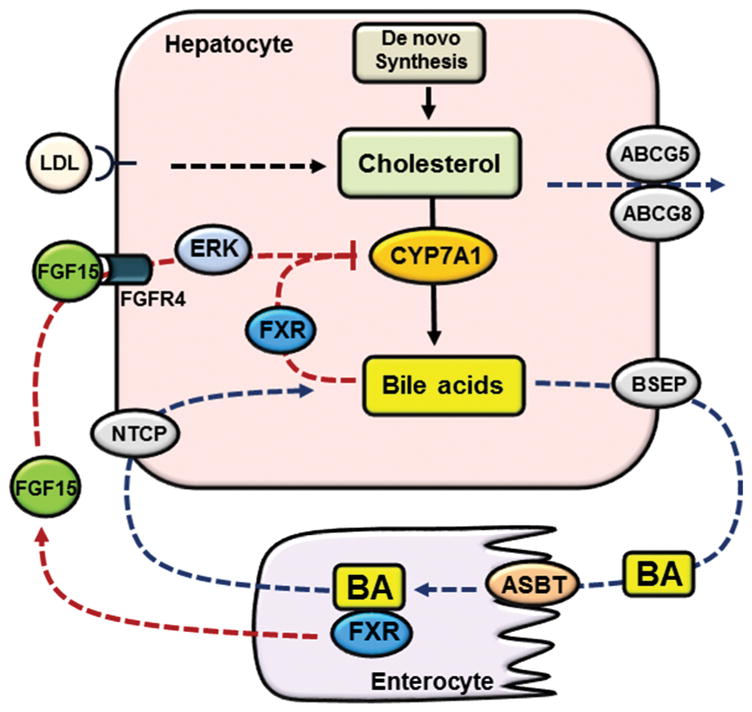
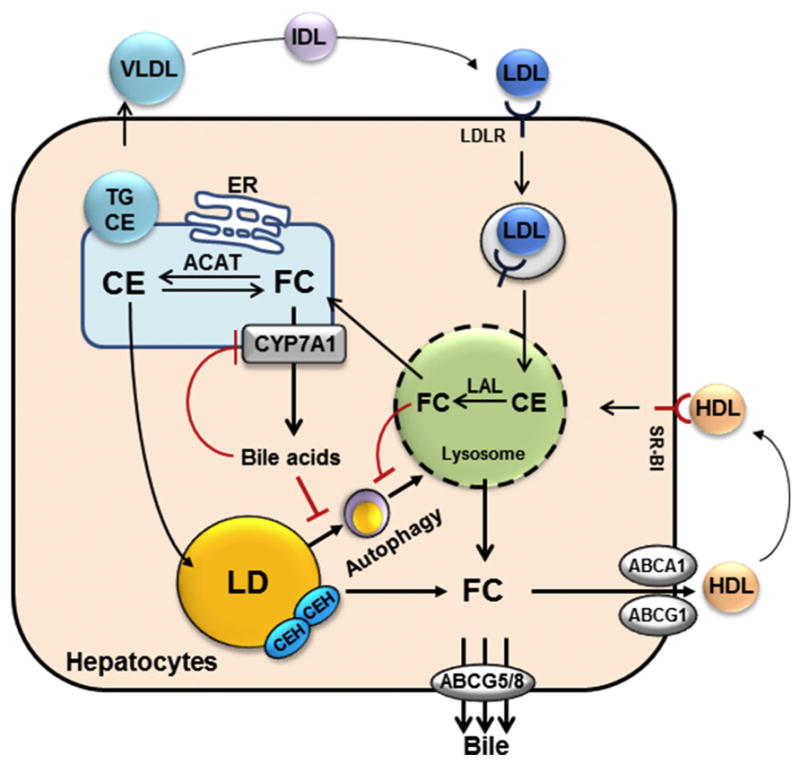
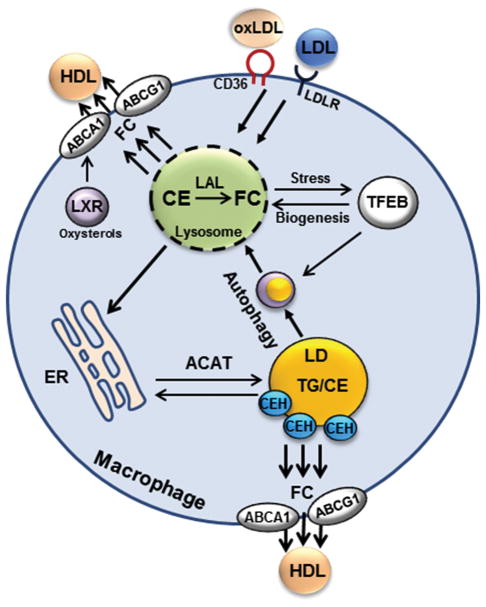
Liver is the major organ that regulates whole body cholesterol metabolism. Disrupted hepatic cholesterol homeostasis contributes to the pathogenesis of nonalcoholic steatohepatitis, dyslipidemia, atherosclerosis, and cardiovascular diseases. Hepatic bile acid synthesis is the major catabolic mechanism for cholesterol elimination from the body. Furthermore, bile acids are signaling molecules that regulate liver metabolism and inflammation. Autophagy is a highly-conserved lysosomal degradation mechanism, which plays an essential role in maintaining cellular integrity and energy homeostasis. In this review, we discuss emerging evidence linking hepatic cholesterol and bile acid metabolism to cellular autophagy activity in hepatocytes and macrophages, and how these interactions may be implicated in the pathogenesis and treatment of fatty liver disease and atherosclerosis.
Keywords: Enterohepatic circulation; Hepatocyte; Hyperlipidemia; Liver injury; Macrophage; Nuclear receptor; Nutrient signaling.
Copyright © 2018. Published by Elsevier B.V.
Conflict of interest statement
Figures
References
-
- Byrne CD, Targher G. NAFLD: A multisystem disease. J Hepatol. 2015;62:S47–S64. - PubMed
-
- Fuchs M, Sanyal AJ. Lipotoxicity in NASH. J Hepatol. 2012;56:291–293. - PubMed
-
- Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, Garcia-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–198. - PubMed
-
- Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, van Bilsen M, Shiri-Sverdlov R, Hofker MH. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48:474–486. - PubMed
-
- Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–1090. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
